
Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling

 , , , and
, , , and
Abstract
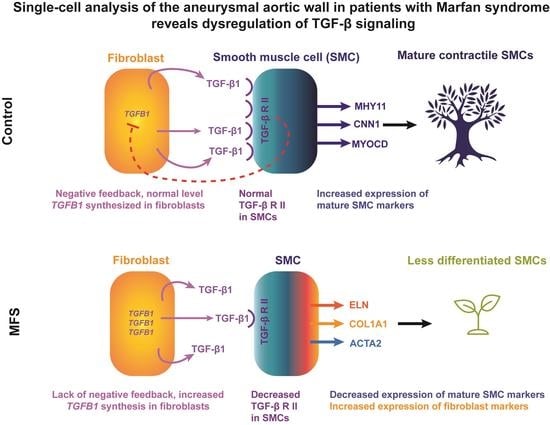
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Tissue Samples
2.2. Antibodies
2.3. Tissue Processing
2.4. Single-Cell RNA Sequencing and Raw Data Processing
2.5. Cell Clustering and Identification
2.6. Differential Gene Analysis and Function Annotation
2.7. Module (Composite) Score
2.8. Correlation Analysis
2.9. Immunofluorescence
3. Results
3.1. Overall Cell Populations and Non-Immune Cells in the Aortic Wall
3.2. Heterogeneous SMC Population in the Aortic Wall
3.2.1. Mature Contractile and Contractile SMCs
3.2.2. Stressed SMCs
3.2.3. Fibromyocytes and Intermediate SMCs
3.2.4. De-Differentiated, Proliferative SMCs
3.3. Fibroblast Phenotypes in the Aortic Wall
3.3.1. Stressed Fibroblasts
3.3.2. Quiescent Fibroblasts
3.3.3. Activated Fibroblasts
3.4. Endothelial Cells and MSCs in the Aortic Wall
3.5. Immune-like Non-Immune Cells in the Aortic Wall
3.6. Phenotypic Continuum of SMCs and Fibroblasts
3.7. Decreased SMC Differentiation in MFS
3.8. Cell-Specific Expression of Canonical TGF-β Pathways among Non-Immune Cell Populations
3.9. Compromised Canonical TGF-β Pathway in Non-Immune Cells in MFS
3.10. Other Key Pathways in Non-Immune Cells in MFS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Ramirez, F.; Caescu, C.; Wondimu, E.; Galatioto, J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFbeta signaling and cell stemness. Matrix Biol. 2018, 71–72, 82–89. [Google Scholar] [CrossRef]
- Robinson, P.N.; Arteaga-Solis, E.; Baldock, C.; Collod-Beroud, G.; Booms, P.; De Paepe, A.; Dietz, H.C.; Guo, G.; Handford, P.A.; Judge, D.P.; et al. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 2006, 43, 769–787. [Google Scholar] [CrossRef] [Green Version]
- Crosas-Molist, E.; Meirelles, T.; Lopez-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko Del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizabal, Y.; et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef] [Green Version]
- Pedroza, A.J.; Koyano, T.; Trojan, J.; Rubin, A.; Palmon, I.; Jaatinen, K.; Burdon, G.; Chang, P.; Tashima, Y.; Cui, J.Z.; et al. Divergent effects of canonical and non-canonical TGF-β signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J. Cell. Mol. Med. 2020, 24, 2369–2383. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Hong, Y.; He, H.; Huang, X.; Tao, W.; Liang, X.; Zhang, Y.; Li, X. TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging 2019, 11, 3574–3584. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Virmani, R.; Majesky, M.W. An update on clonality: What smooth muscle cell type makes up the atherosclerotic plaque? F1000Res 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.; Maechler, M. Matrix: Sparse and Dense Matrix Classes and Package Version 1.2–18. Available online: https://CRAN.R-project.org/package=Matrix (accessed on 8 September 2021).
- Bengtsson, H. Functions That Apply to Rows and Columns of Matrices (and to Vectors). R Package Version 0.55.0. Available online: https://CRAN.R-project.org/package=matrixStats (accessed on 8 September 2021).
- Kolde, R. Pretty Heatmaps. R Package Version 1.0.12. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 8 September 2021).
- R Core Team. A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 8 September 2021).
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. R Package Version 1.4.0. Available online: https://CRAN.R-project.org/package=stringr (accessed on 8 September 2021).
- Wickham, H.; Francois, R.; Henry, L.; Muller, K. Dplyr: A Grammar of Data Manipulation. R Package Version 0.8.4. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 8 September 2021).
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Risso, D.; Perraudeau, F.; Gribkova, S.; Dudoit, S.; Vert, J.P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 2018, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Morrow, D.; Guha, S.; Sweeney, C.; Birney, Y.; Walshe, T.; O’Brien, C.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch and vascular smooth muscle cell phenotype. Circ. Res. 2008, 103, 1370–1382. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; Campbell, K.R.; Zhang, A.W.; Kabeer, F.; Lim, J.L.P.; Biele, J.; Eirew, P.; Lai, D.; McPherson, A.; Kong, E.; et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 2019, 20, 210. [Google Scholar] [CrossRef] [Green Version]
- Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. The role of osteoprotegerin in the crosstalk between vessels and bone: Its potential utility as a marker of cardiometabolic diseases. Pharmacol. Ther. 2018, 182, 115–132. [Google Scholar] [CrossRef]
- Coppock, D.L.; Kopman, C.; Scandalis, S.; Gilleran, S. Preferential gene expression in quiescent human lung fibroblasts. Cell Growth Differ. 1993, 4, 483–493. [Google Scholar]
- Marthandan, S.; Priebe, S.; Hemmerich, P.; Klement, K.; Diekmann, S. Long-term quiescent fibroblast cells transit into senescence. PLoS ONE 2014, 9, e115597. [Google Scholar] [CrossRef] [Green Version]
- Pollina, E.A.; Legesse-Miller, A.; Haley, E.M.; Goodpaster, T.; Randolph-Habecker, J.; Coller, H.A. Regulating the angiogenic balance in tissues. Cell Cycle 2008, 7, 2056–2070. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.; Hughes, M.; Krishnamoorthy, S.; Zou, S.; Zhang, L.; Wu, D.; Zhang, C.; Curci, J.A.; Coselli, J.S.; Milewicz, D.M.; et al. Critical role of ADAMTS-4 in the development of sporadic aortic aneurysm and dissection in mice. Sci. Rep. 2017, 7, 12351. [Google Scholar] [CrossRef] [Green Version]
- Lemons, J.M.; Feng, X.J.; Bennett, B.D.; Legesse-Miller, A.; Johnson, E.L.; Raitman, I.; Pollina, E.A.; Rabitz, H.A.; Rabinowitz, J.D.; Coller, H.A. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010, 8, e1000514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manabe, I.; Shindo, T.; Nagai, R. Gene expression in fibroblasts and fibrosis: Involvement in cardiac hypertrophy. Circ. Res. 2002, 91, 1103–1113. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, S.; Marrow, S.M.W.; Watch, L.A.; Young, P.P. Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nat. Commun. 2019, 10, 3027. [Google Scholar] [CrossRef] [PubMed]
- Domenga, V.; Fardoux, P.; Lacombe, P.; Monet, M.; Maciazek, J.; Krebs, L.T.; Klonjkowski, B.; Berrou, E.; Mericskay, M.; Li, Z.; et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004, 18, 2730–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Chen, S.Y. Transforming growth factor-β and smooth muscle differentiation. World J. Biol. Chem. 2012, 3, 41–52. [Google Scholar] [CrossRef]
- Neptune, E.R.; Frischmeyer, P.A.; Arking, D.E.; Myers, L.; Bunton, T.E.; Gayraud, B.; Ramirez, F.; Sakai, L.Y.; Dietz, H.C. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003, 33, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Mummery, C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 2000, 44, 253–265. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.H.; Zhang, L.; Ren, P.; Nguyen, M.T.; Zou, S.; Wu, D.; Wang, X.L.; Coselli, J.S.; LeMaire, S.A. AKT2 confers protection against aortic aneurysms and dissections. Circ. Res. 2013, 112, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Pedroza, A.J.; Tashima, Y.; Shad, R.; Cheng, P.; Wirka, R.; Churovich, S.; Nakamura, K.; Yokoyama, N.; Cui, J.Z.; Iosef, C.; et al. Single-cell transcriptomic profiling of vascular smooth muscle cell phenotype modulation in Marfan syndrome aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2195–2211. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Guo, D.C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef]
- Franken, R.; den Hartog, A.W.; de Waard, V.; Engele, L.; Radonic, T.; Lutter, R.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Zwinderman, A.H.; et al. Circulating transforming growth factor-β as a prognostic biomarker in Marfan syndrome. Int. J. Cardiol. 2013, 168, 2441–2446. [Google Scholar] [CrossRef] [PubMed]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.L.; Yang, J.H.; Song, S.H.; Kim, J.Y.; Jang, S.Y.; Kim, J.M.; Kim, J.A.; Sung, K.I.; Kim, Y.W.; Suh, Y.L.; et al. Positive correlation between the dysregulation of transforming growth factor-beta1 and aneurysmal pathological changes in patients with Marfan syndrome. Circ. J. 2013, 77, 952–958. [Google Scholar] [CrossRef] [Green Version]
- Matt, P.; Schoenhoff, F.; Habashi, J.; Holm, T.; Van Erp, C.; Loch, D.; Carlson, O.D.; Griswold, B.F.; Fu, Q.; De Backer, J.; et al. Circulating transforming growth factor-β in Marfan syndrome. Circulation 2009, 120, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Lindner, V.; Booth, C.; Prudovsky, I.; Small, D.; Maciag, T.; Liaw, L. Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am. J. Pathol. 2001, 159, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Takefuji, M.; Ngai, C.Y.; Carvalho, J.; Bayer, J.; Wietelmann, A.; Poetsch, A.; Hoelper, S.; Conway, S.J.; Mollmann, H.; et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 2016, 118, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- Tieu, B.C.; Ju, X.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A., 3rd; Brasier, A.R.; Tilton, R.G. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J. Vasc. Res. 2011, 48, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.Q.; Geng, L.; Prakash, S.K.; Cao, J.M.; Guo, S.; Villamizar, C.; Kwartler, C.S.; Peters, A.M.; Brasier, A.R.; Milewicz, D.M. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2172–2179. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawson, A.; Li, Y.; Li, Y.; Ren, P.; Vasquez, H.G.; Zhang, C.; Rebello, K.R.; Ageedi, W.; Azares, A.R.; Mattar, A.B.; et al. Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes 2022, 13, 95. https://doi.org/10.3390/genes13010095
Dawson A, Li Y, Li Y, Ren P, Vasquez HG, Zhang C, Rebello KR, Ageedi W, Azares AR, Mattar AB, et al. Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes. 2022; 13(1):95. https://doi.org/10.3390/genes13010095
Chicago/Turabian StyleDawson, Ashley, Yanming Li, Yang Li, Pingping Ren, Hernan G. Vasquez, Chen Zhang, Kimberly R. Rebello, Waleed Ageedi, Alon R. Azares, Aladdein Burchett Mattar, and et al. 2022. "Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling" Genes 13, no. 1: 95. https://doi.org/10.3390/genes13010095
APA StyleDawson, A., Li, Y., Li, Y., Ren, P., Vasquez, H. G., Zhang, C., Rebello, K. R., Ageedi, W., Azares, A. R., Mattar, A. B., Sheppard, M. B., Lu, H. S., Coselli, J. S., Cassis, L. A., Daugherty, A., Shen, Y. H., & LeMaire, S. A. (2022). Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes, 13(1), 95. https://doi.org/10.3390/genes13010095