1. Introduction
Tilapia comprises over 100 species of freshwater fish in Africa that belong to the genera
Oreochromis,
Sarotherodon, and
Tilapia. Seventy-five countries have economies of scale for feeding Nile tilapia. Tilapia is one of the most important economic farmed fish in Taiwan, as well as in the world. According to the Food and Agriculture Organization (FAO), tilapia is one of the important species for providing protein in terms of the human diet [
1]. Selective breeding programs have been widely used in aquatic farming (more than 60 species) for fish such as common carp, grass carp, rainbow trout, Atlantic salmon, channel catfish, sea bream, oyster, shrimp, and tilapia [
2]. The GIFT strain (Genetic Improvement of Farmed Tilapia) in Nile tilapia (
Oreochromis niloticus) started in 1989 and originated from the GIFT selective breeding project in the Philippines [
3]. While the GIFT strain is well known for its superior growth performance, it is characterized by a comparatively weakened immune system [
4].
Diseases associated with Nile tilapia have gathered serious attention in recent years, including viral encephalitis of tilapia larvae (primary characterization of a novel herpes-like virus) and TLEV (tilapia larva encephalitis virus) [
5,
6]. Many Gram-positive and Gram-negative bacteria can infect tilapia, such as of the following genera:
Aeromonas, Citrobacter, Edwardsiella, Flavobacterium, Pseudomonas, Streptococcus, and
Mycobacterium [
6,
7]. Among these,
Streptococcus agalactiae and
Streptococcus iniae are the major bacterial pathogens in tilapia [
8]. To solve problems of the related diseases, research has sought to improve the immune capacity of fish through the upregulation expression of antimicrobial peptides (AMPs) [
9]. AMP expression is one mechanism used by the innate immune system of fish [
10] that can effectively inhibit the growth of bacteria, and even improved survival after the injection of synthetic AMPs has been observed [
11]. In fish, most AMPs are α-helical peptides [
12]. Moreover, a total of 122 fish AMPs have been reported, including cathelicidin, chryosophsin, dicentracin, epinecidine, hepcidin, misgurin, oncorhynsin, and piscidin [
13,
14,
15,
16]. In contrast to blue tilapia (
Oreochromis aureus), Nile tilapia has stronger disease resistance [
17]. Another study also revealed that Nile tilapia had more AMP genes than blue tilapia (
Oreochromis aureus), especially hepcidin [
18]. Additionally, gene diversification and amplification processes have occurred in fish hepcidin [
19]. These results suggest a correlation between disease resistance and AMPs.
Hepcidin is a regulatory factor of iron and also a hepatic antimicrobial peptide (HAMP) that is primarily expressed in the liver [
20,
21]. It has been extensively studied in various species, including fish. Three hepcidins have been found in
Oreochromis mossambicus, namely Th1-5, Th2-2, and Th2-3 [
21]. Two AMPs, Hep-JF1 and Hep-JF2, were found in Japanese flounder (
Paralichthys olivaceus). Six hepcidin genes (LcHamps) with diversified regulation and functions involved in antibacterial activity, antiviral activity, and regulation of intracellular iron metabolism were identified in large yellow croaker (
Larimichthys crocea) genome [
19]. Hepcidin showed the strongest expression in the liver after LPS induction on the convict cichlid (
Archocentrus nigrofasciatus) [
11]. Studies of pathogenic bacteria treated with synthetic hepcidin peptides showed that the pathogen cell membranes were destroyed. Studies also indicated that oral intake or injection of hepcidin in zebrafish could inhibit bacterial growth and stimulate the host immune response, which significantly improved their survival rate following pathogen infection [
22,
23]. Hepcidin was used against
Staphylococcus aureus and
Vibrio vulnificus in spotted grouper (
Epinephelus coioides) [
24]. Pagaporn et al. [
25] mentioned that hepcidin could inhibit both virulence and growth of bacteria by reducing the level of metals (e.g., Fe) in fish tissues. The study indicated that hepcidin enhanced streptococcosis resistance in Nile tilapia.
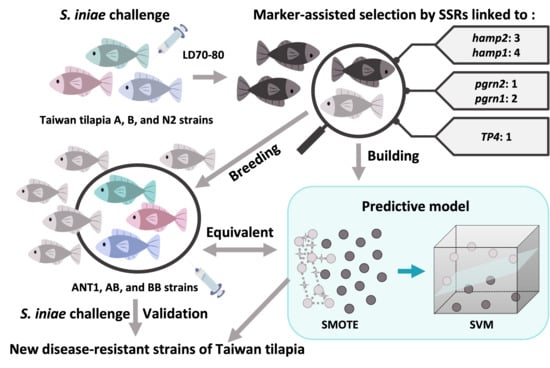
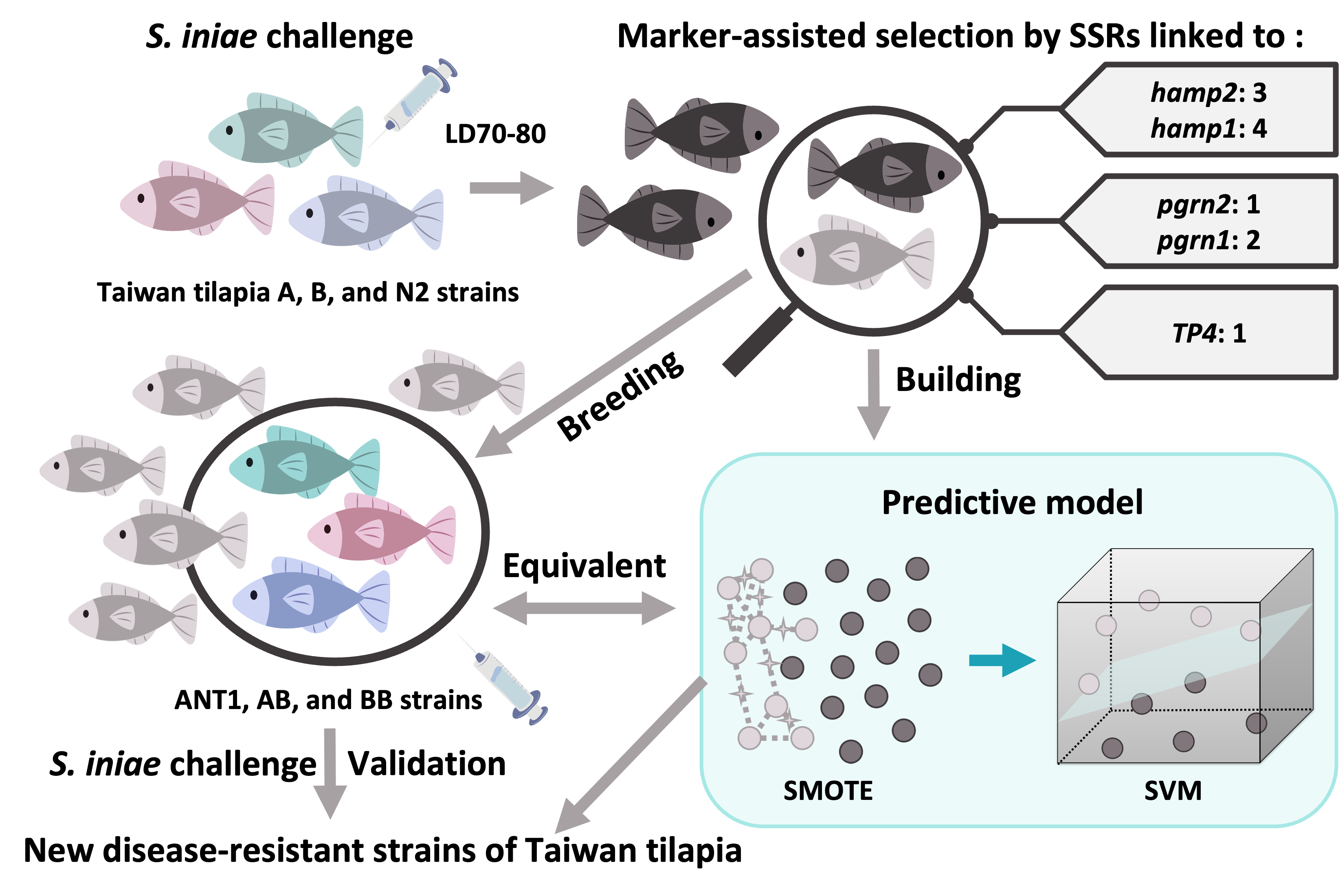
Simple sequence repeats (SSRs), also known as microsatellites or short tandem repeats (STRs), are tandem repeats of short sequence motifs commonly found in eukaryotic genomes. They are also considered as useful genetic markers for genetic diversity analysis, DNA fingerprinting, and linkage mapping. Several studies have shown that the distribution of SSRs in the genome is non-random. There are more SSRs in the untranslated regions (UTRs) than in the coding regions and are thought to be associated with the regulation of gene expression [
26]. Recent studies have revealed that microsatellite polymorphisms on a genome-wide scale contribute to the heritability of human gene expression. Evidence has been found for the role of microsatellites in regulating transcription factor binding, methylation, promoter, enhancers, mRNA stability, alternative splicing, nucleosome modification, and noncoding RNA [
27].
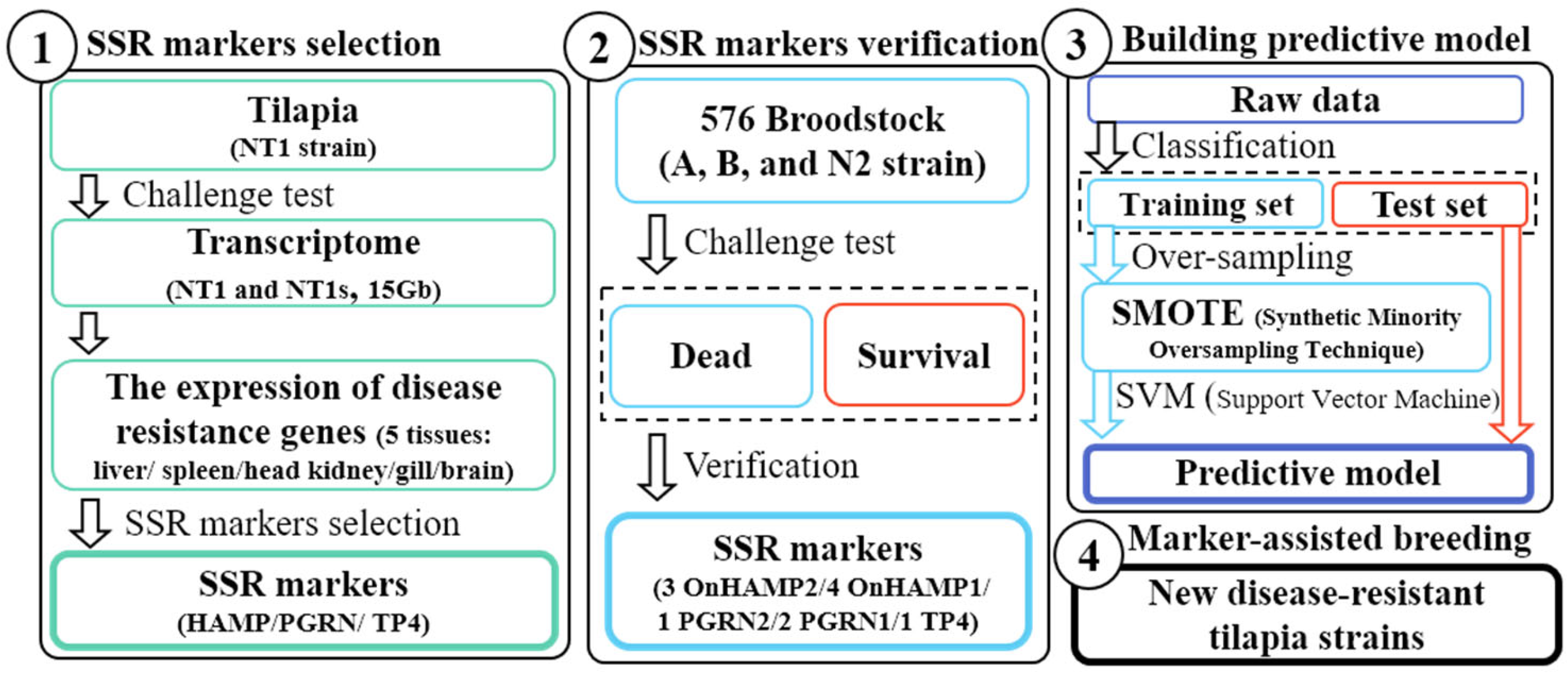
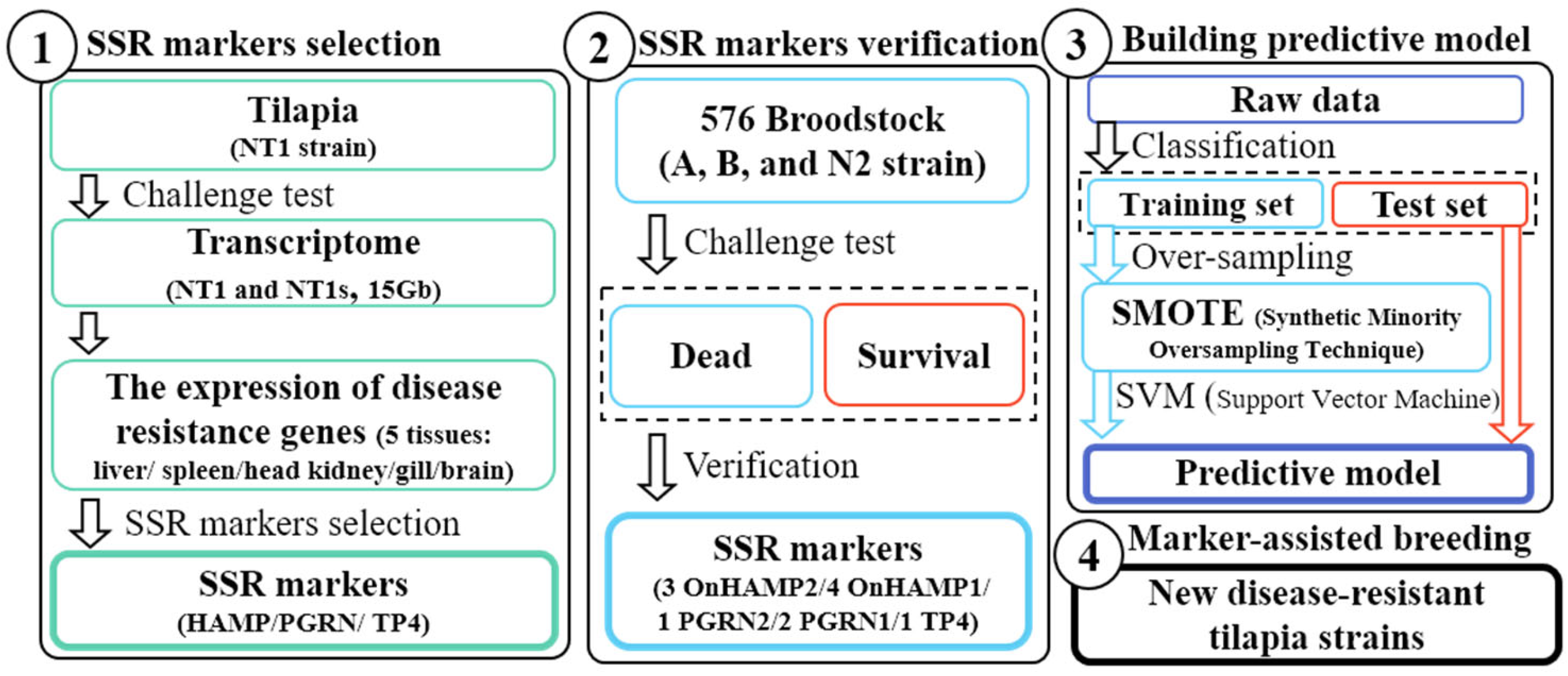
With the development of genetic technology, marker-assisted selection has been applied in many aquaculture species [
28]. However, research developing microsatellite markers of disease resistance for fish is scarce. In this study, disease-resistance-associated microsatellite markers were developed from the Nile tilapia genome. To confirm that the microsatellite markers were associated with disease resistance, seven populations were chosen and analyzed by
Streptococcus infection. As far as we know, this is the first report describing the development of disease-resistance-associated microsatellites in Taiwan tilapia. Thus, we hope our findings will have a positive practical impact on the tilapia industry in Taiwan.
4. Discussion
At the beginning of this study, the NT1 strain was infected by
S. iniae 89353 (10
4 cfu/g) at 12 hpi for the collection of the transcriptome sample. In contrast to previous research [
49,
50], a lower dose and longer response time were selected to allow observation of the recovery response after challenging with
S. iniae. Even if there was no enormous differential gene expression in the transcriptome result (
Table S4), the qPCR results indicate that
hamp gene expression increased substantially.
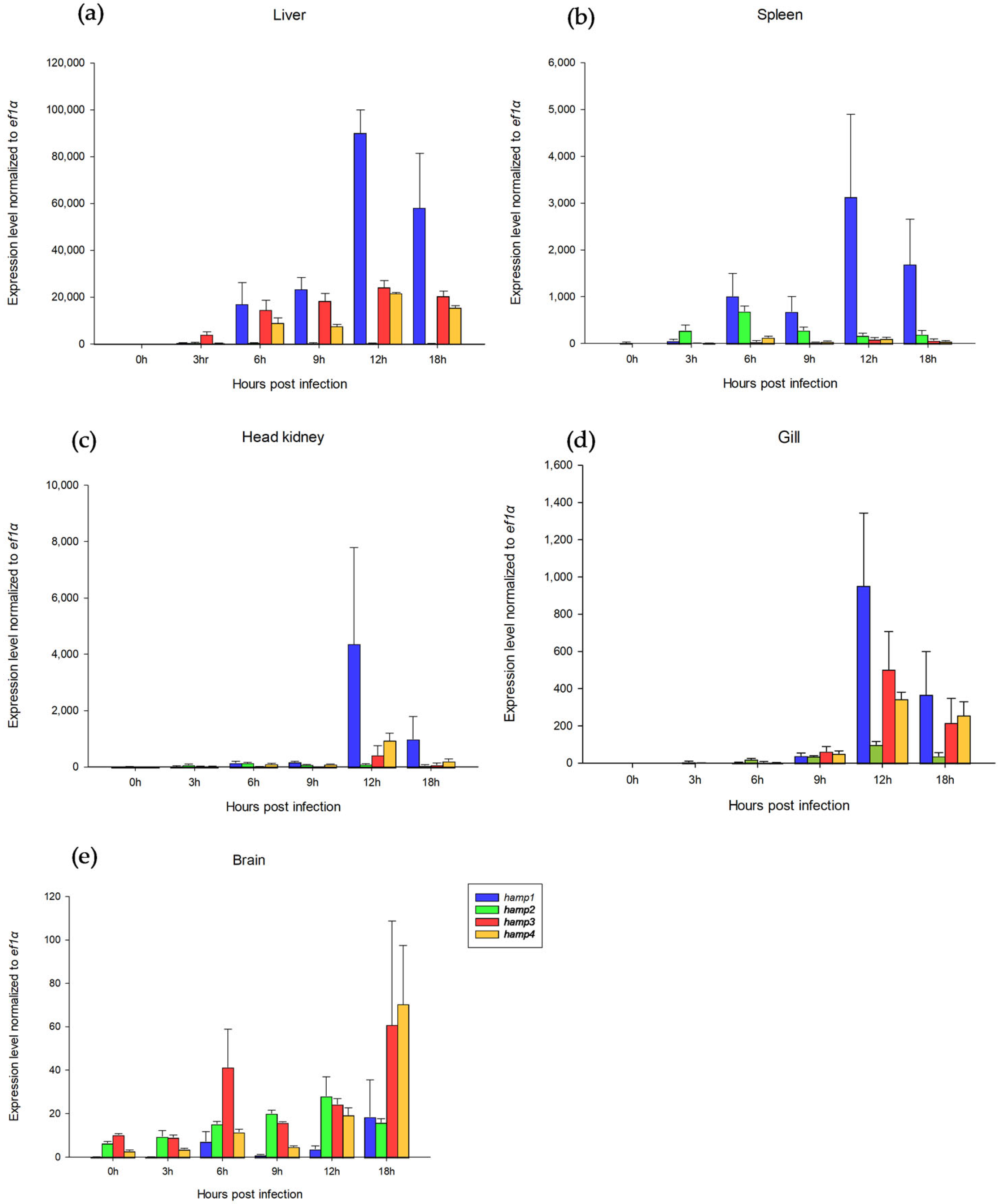
Figure 4 shows the
hamp1,
hamp3, and
hamp4 levels reaching a maximum at 12 h (
hamp2 at 3–6 h) in all tissues during
S. iniae infection. It was also reported that hepcidin expression increased remarkably after pathogenic infection [
51,
52].
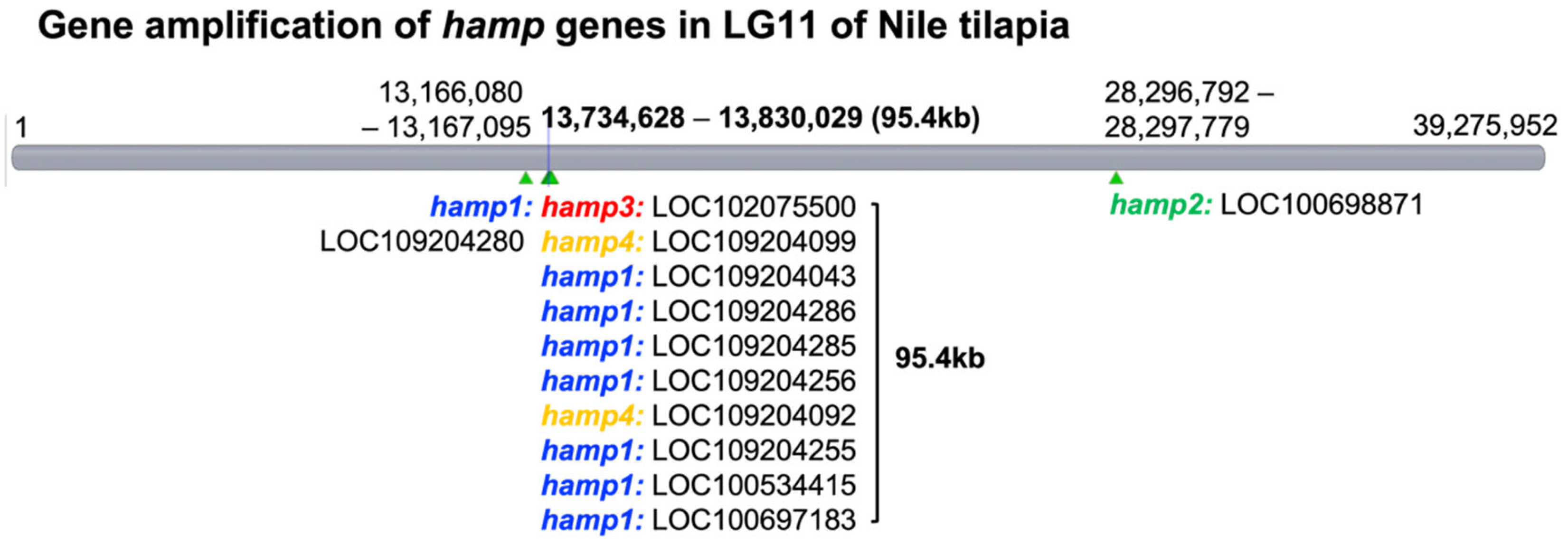
Figure 2 and
Figure 3 present the results for 12 hepcidin genes in Nile tilapia. These results are similar to previous reports showing that multiple hepcidin gene copies have been generated through duplication and diversification processes in fish [
19,
53,
54,
55], and that Nile tilapia has more hepcidin genes than blue tilapia [
18]. These results suggest that hepcidin gene amplification is associated with disease resistance in tilapia. These genes were found to be significantly upregulated after the challenge experiment. Surprisingly, hepcidin gene amplification was also observed (
Figure 2 and
Figure 3). It is therefore assumed that these genes play an important role in the infective response. Furthermore, the short product of
pgrn could enhance disease resistance via participation in the regulation of innate immune-related genes in tilapia [
56]. The GRN-41 peptide, which is product of
Pgrn1 generated by alternative RNA splicing, also has antimicrobial activity against
Vibrio [
57]. The disease resistance of tilapia can be effectively increased by TP4 (tilapia piscidin 4) [
58,
59,
60,
61].
The correlation analysis by chi-square test (
Table 2,
Table 3 and
Table 4) and Z-test (
Table S10) suggest the relationship between SSRs polymorphism and disease resistance. Moreover,
Figure 4 indicates the association of gene expression and disease resistance in HAMP. We hypothesized that different SSRs lengths affect gene expression, which in turn caused effects on disease resistance. The effect of SSRs on gene expression has been reported [
26,
27]. Some of the examples of SSRs affecting gene expression are as follows. In human, there was a long CGG trinucleotide repeat in the 5′-UTR of the
FMR1 gene. This SSR was adjacent to the promoter and affected the performance of the
FMR1 gene, leading to fragile X syndrome (FXS) [
62]. In another study, the (GA)n microsatellite sequence of the promoter region has been shown to bind the GAGA factors (GAF) proteins in
Drosophila GAGA factor (GAF) [
63,
64]. The GAF is a multifunctional protein that influences gene expression, the communication between promoters and enhancers, nucleosome organization, and chromosome structure [
65]. In mammalian cells, the transcription start site (TSS) at the 5′-UTR end of the promoter is affected by GAF binding sequences [
66]. Streelman and Kocher present the (CA)n microsatellite which is found in the prolactin 1 (
prl 1) gene of the 5′-UTR is associated
prl 1 gene expression in tilapia [
67]. Both
prl1 and growth hormone (GH) gene polymorphism has been proven to be linked to growth in tilapia [
68]. Overall, our study is based on these disease-resistance-related genes of tilapia, and we selected samples for intraperitoneal injection of the highly pathogenic
S. iniae. Developing microsatellites associated with these antimicrobial peptides might effectively aid in the marker-assisted selection of disease-resistant strains of aquaculture species.
In the above-mentioned study, we concluded that SSR and disease resistance are indeed associated, but the results of these markers are not similar to different strains. As the result, the correlation analyses of A, B, and N2 populations were, respectively, 10, 1, and 3 of the 11 microsatellite markers after the correlation analysis between genotype and survival by the chi-square test (
p < 0.05). Then, there were four microsatellite markers (SSR4, 5, 7, 19) in the B strain and five (SSR2, 14, 17, 19, 22) in the N2 strain (
Table S10), both of which showed a statistically significant difference (
p < 0.05) when analyzing the association between all genotypes of each SSR and the numbers in the alive or dead groups by Z-test. These results indicate that the disease resistance of strain A is higher than in strains B and N2, and B and N2 strains are similar. The direct reason may be the variation in disease resistance of different strains. This result is also consistent with the result of the
S. iniae challenge experiment (the LD71.3 of strain A was 2 × 10
6 CFU/g, the LD73.2 of strain B was 6 × 10
5 CFU/g, and the LD79.7 of strain N2 was 6.5 × 10
5 CFU/g).
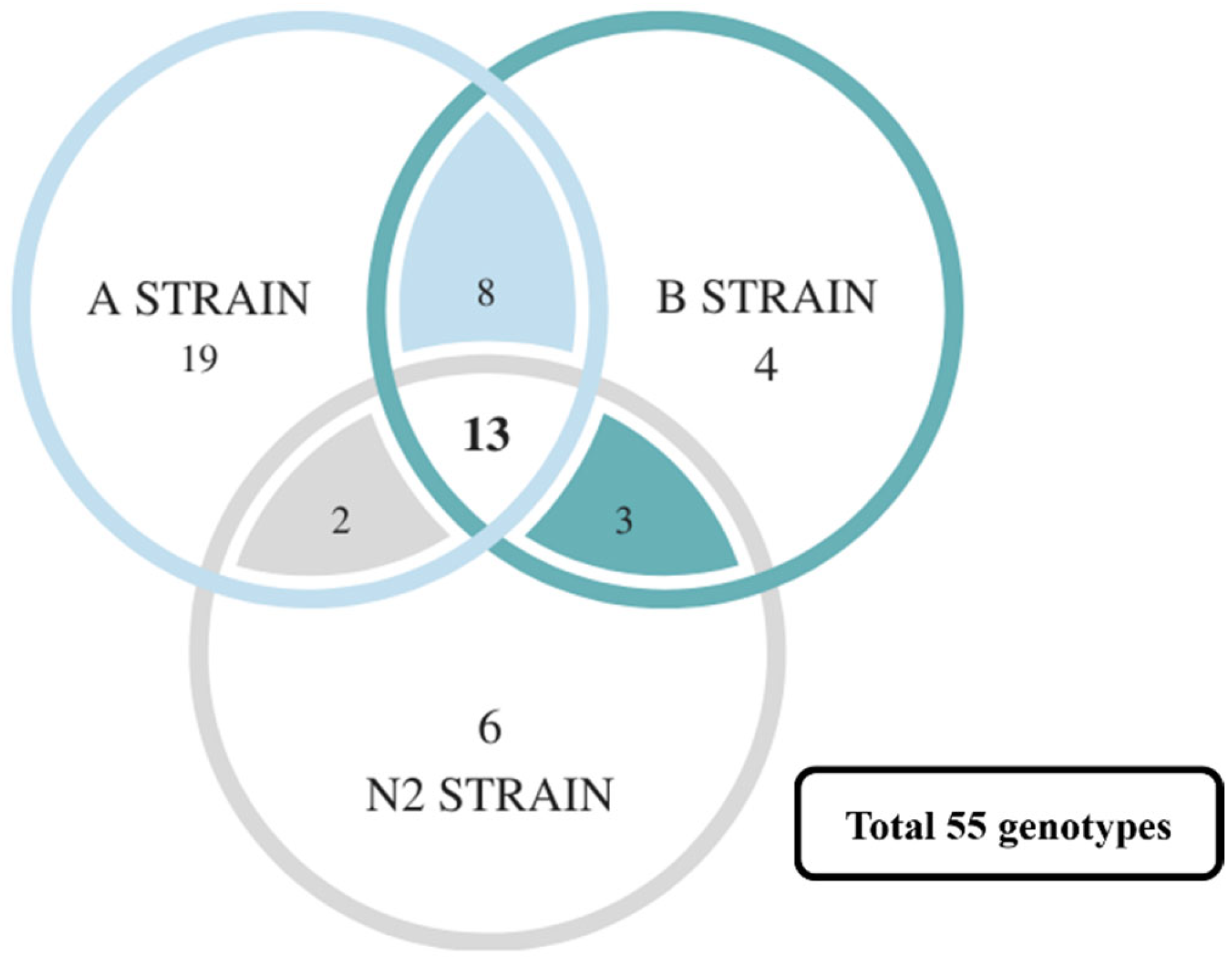
Furthermore, the correlation analysis results of all SSRs in the A, B, and N2 strains were compared. A total of 271 genotypes were found, of which 55 genotypes were related to survival (
Table S10). However, many survival-related genotypes were only found in specific strains (such as SSR5, 8, and 21), even having a statistically significant difference (especially strain A,
Figure 6). This may because of the following: (1) the number of genotypes is too large, but the number of fish related to each genotype is too small; or (2) the challenge experiments with a high lethal dose create a smaller number of survivor groups. Nevertheless, the SSR5, SSR8, SSR19, and SSR22 in strain A are still significantly related to survival. It was found that the number and proportion of most genotypes that have no significant difference in other groups were still associated with survival via a comparison with the number and proportion of all microsatellite markers in the death and survival groups (
Table S10). Fuji et al. [
69] mentioned that, despite there being no significant correlation in the first generation of statistics, the ratio of certain genotypes could be increased by repeated backcrossing; thus, a microsatellite closely linked to lymphocystis disease resistance (LD-R) was selected from 50 microsatellites. They also attempted to transfer the closely linked LD-R microsatellite marker Poli9-8TUF into a commercial strain, and they successfully developed a new disease-resistant strain [
70]. These findings were corroborated in our study; although some genotypes are only significantly associated with survival in specific strains, they are nevertheless associated with survival.
Although 55 disease-related genotypes were found by a Z-test, most genotypes were found in strain A. It was proposed that more disease-resistant markers could be found in the strain demonstrating broader disease resistance. However, those with both significant differences and no significant differences might still be potential molecular markers. As mentioned in the previous section, even if 37 genotypes only have a significant difference in strain A, the number and proportion of most genotypes which have no significant difference in other strains are still associated with survival. Moreover, disease resistance may also be graded according to different combinations of genotypes. Our hypotheses are the following: (1) every genotype has different strengths; and (2) different combinations of genotypes cause variations in effectiveness. To address the challenges arising from this variation, predictive modeling was built by strains A and B.
Table 6 shows the evaluation values of our predictive model. Accuracy is the factor most commonly evaluated for a predictive model and is represented by a value derived from the number of samples of correct judgment (true positive and true negative) divided by all samples [
71]. Accuracy was 0.8598 in the training set and 0.8462 in the testing set (range from 0 to 1; the closer to 1, the better). However, this accuracy does not apply when the actual number of positive samples is low; thus, precision and sensitivity are used. Precision and sensitivity are both concerned with a true positive, but from different perspectives. Precision relies on predicting the actual precision in a positive situation, while sensitivity predicts “how much” of the actual positive answer can be recalled in a positive situation [
45]. The precision in the training set was 0.7883 and the sensitivity was 0.9837. The precision in the testing set was 0.8387 and the sensitivity was 0.9630. Both values were close to 1 (especially the sensitivity). The
F1-score is the harmonic average of the two values (precision and sensitivity) and is often used to evaluate the accuracy of a given model [
45]. The
F1-scores were 0.8752 and 0.8966 for the training and test sets, respectively (range from 0 to 1; the closer to 1, the better). The sensitivity denotes detection of how many samples will actually die. The specificity represents the number of samples that survived via predictive model detection. The higher the sensitivity and specificity values, the better the model in terms of prediction (range from 0 to 1; the closer to 1, the better) [
72]. The specificity for the training set and the test set was 0.7358 and 0.5833, respectively, which indicated that the actual surviving samples in the test set were slightly lower than in the training set. MCC is usually regarded as a balanced indicator. In essence, MMC is a correlation coefficient that describes the actual classification and the predicted classification. The range is from −1 to 1. A value of 1 describes a perfect prediction, a value of 0 shows that the predicted result is worse than the random result, and −1 demonstrates that the predicted classification and the actual classification are completely inconsistent [
46]. The MCC in the training set and testing set was 0.7427 and 0.6244, respectively. Additionally, the closer the FPR, FDR, and FNR are to 0, the better. The results showed that only the FPR of the testing set was higher. This is because FPR = 1 − specificity, and a lower correct number of tested actual surviving samples means a higher incorrect number of tested surviving samples. Therefore, we also plotted the ROC curve (
Figure S1) using false positive rate (FPR) and true positive rate (TPR). If the ROC curve is equal to the diagonal line, the model shows no discrimination. If the ROC curve moves to the upper left corner, the model is more sensitive to disease resistance (the lower false positive rate), which means the model has better discrimination. Meanwhile, the ROC curve is used to calculate the area under curve, which ranges from 0 to 1; the larger value, the better [
44,
45,
47]. The AUC was 0.983 in the training set, which meant excellent discrimination. The AUC was 0.849 in the testing set, which meant good discrimination. Combining the evaluation values from
Table 6 and
Figure S1, it is proposed that this model has certain credibility in predicting the mortality of a population.
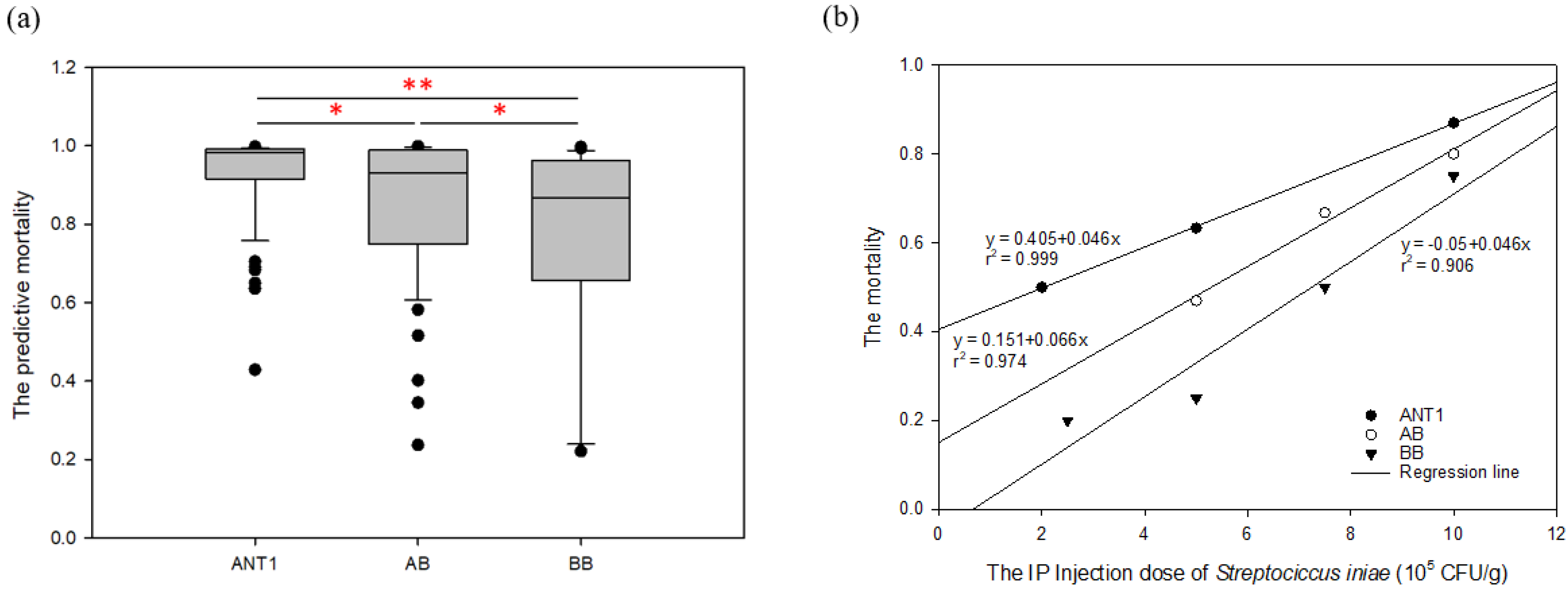
The model was applied to detect the new
Streptococcus-resistant group (F
1). The result reveals that the proportions of predictive death numbers in the ANT1, AB, and BB groups were 95/96 (0.99), 52/55 (0.945), and 31/40 (0.775), respectively. Meanwhile, the mean predictive mortality (±SEM) of ANT1 was 0.932 ± 0.011, AB was 0.861 ± 0.026, and BB was 0.765 ± 0.041 (
Table S10). Moreover, there are more samples with lower mortality in the BB group (
Figure 5a). As a result, the trend of predictive disease resistance (
Figure 5a) corresponded with actual disease resistance (
Figure 5b). The mortality of ANT1 was highest, next was AB; BB was the lowest group. The predictive model, which was built by disease-resistance-associated microsatellites, could not only predict the mortality of a pure line but also of hybrid offspring. These results may indicate that the resistance-related genotypes which are found from F
0 are still applicable in the offspring. Moreover, this predictive model can also estimate the mortality rate in genotype combinations. There are still numerous issues with this model: (1) the
S. iniae dose does not ensure mortality and only an approximate relative value; (2) offspring not sharing the same genotype as their parent leads to inaccurate interpretation; and (3) the sample size may be too small to build a predictive model. Incorporating more information will increase the accuracy of predictions. To establish a precise predictive model, the dose of the challenge experiment and the time of death should be added to this SVM model, or different models should be established in further experiments. In addition, the predictive model could also be strengthened via machine learning during the breeding process.
With the development of molecular biotechnology, a huge variety of genome-based biotechnologies have been applied to the field of aquaculture research. However, most Nile tilapia breeding relies on traditional breeding methods to select phenotypes, such as growth rate, weight, and length. Relative to the aquaculture industry, modern genome-based strategies (e.g., marker-assisted selection and genomic selection breeding) have been widely using in agriculture and animal industries. Even though marker-assisted selection has only begun to be applied in the aquaculture industry in recent years, some cases of aquaculture studies can be found; for instance, high growth rate, cold resistance, and disease resistance in flatfish [
69,
70], rainbow trout [
73,
74,
75,
76,
77], and carp [
78,
79]. Thus far, most of the current research of marker-assisted selection has focused on developing massive SNPs, SSRs, and deletions in Nile tilapia [
80] for sex determination [
81], population structure analysis [
82], improvement of growth and fillet yield [
83,
84], and cold stress [
85]. Often, few markers are used in breeding, which is not only time consuming but expensive. Overall, there are still only a few areas of research into disease-resistance-associated microsatellites in tilapia, but all commercial tilapia strains in Taiwan are hybridized. Previous studies may therefore not provide precise information directly relevant for the Taiwan tilapia industry.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}