

Integrative System Biology Analysis of Transcriptomic Responses to Drought Stress in Soybean (Glycine max L.)



Abstract
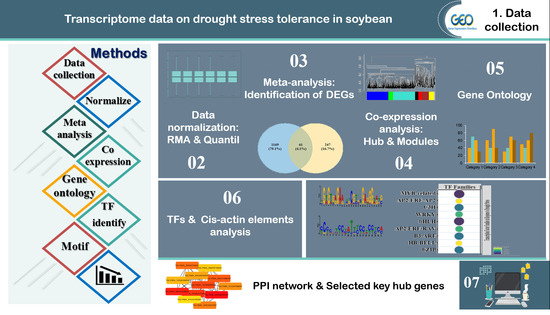
:
1. Introduction
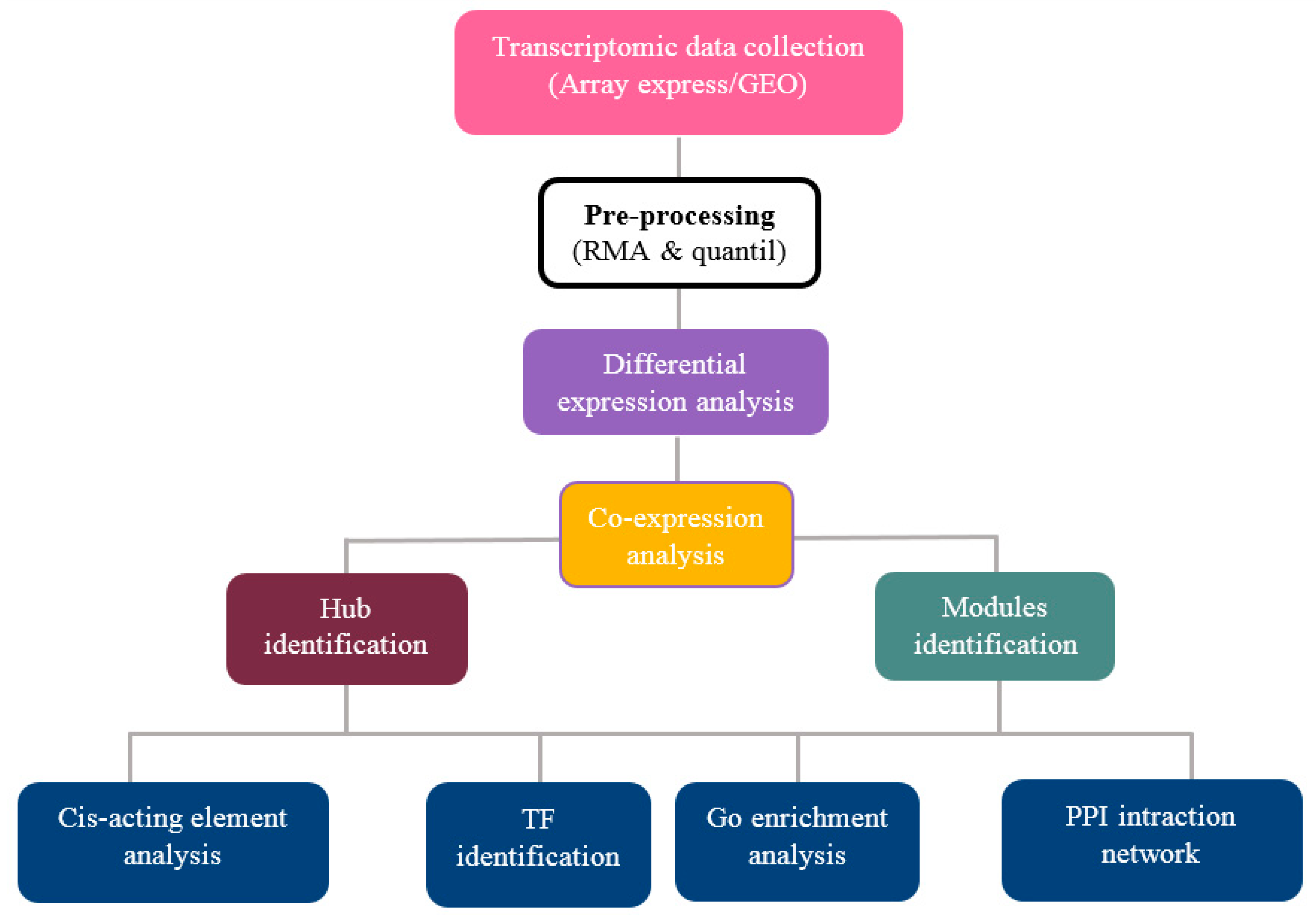
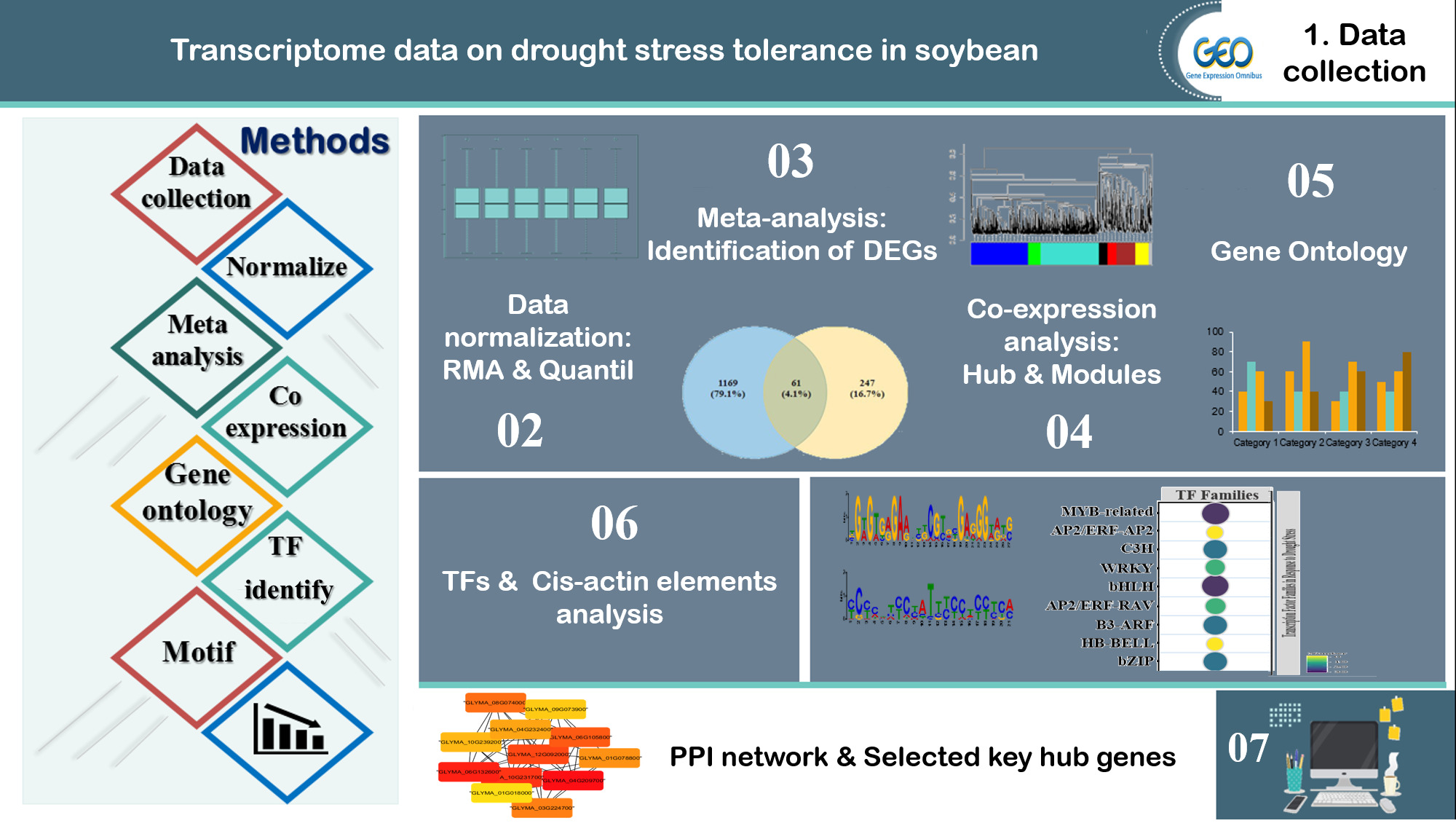
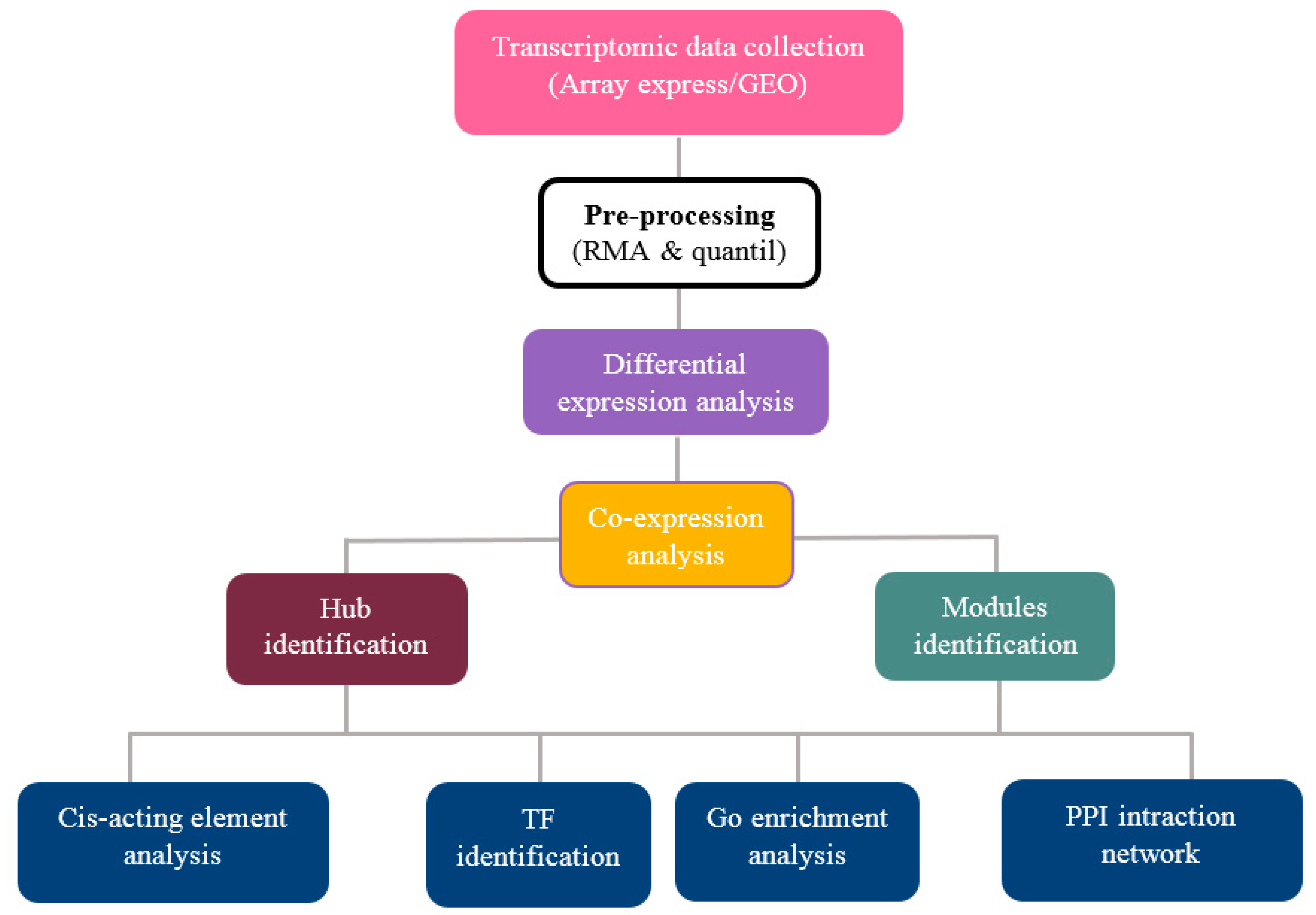
2. Materials and Methods
2.1. Data Collection and Pre-Processing
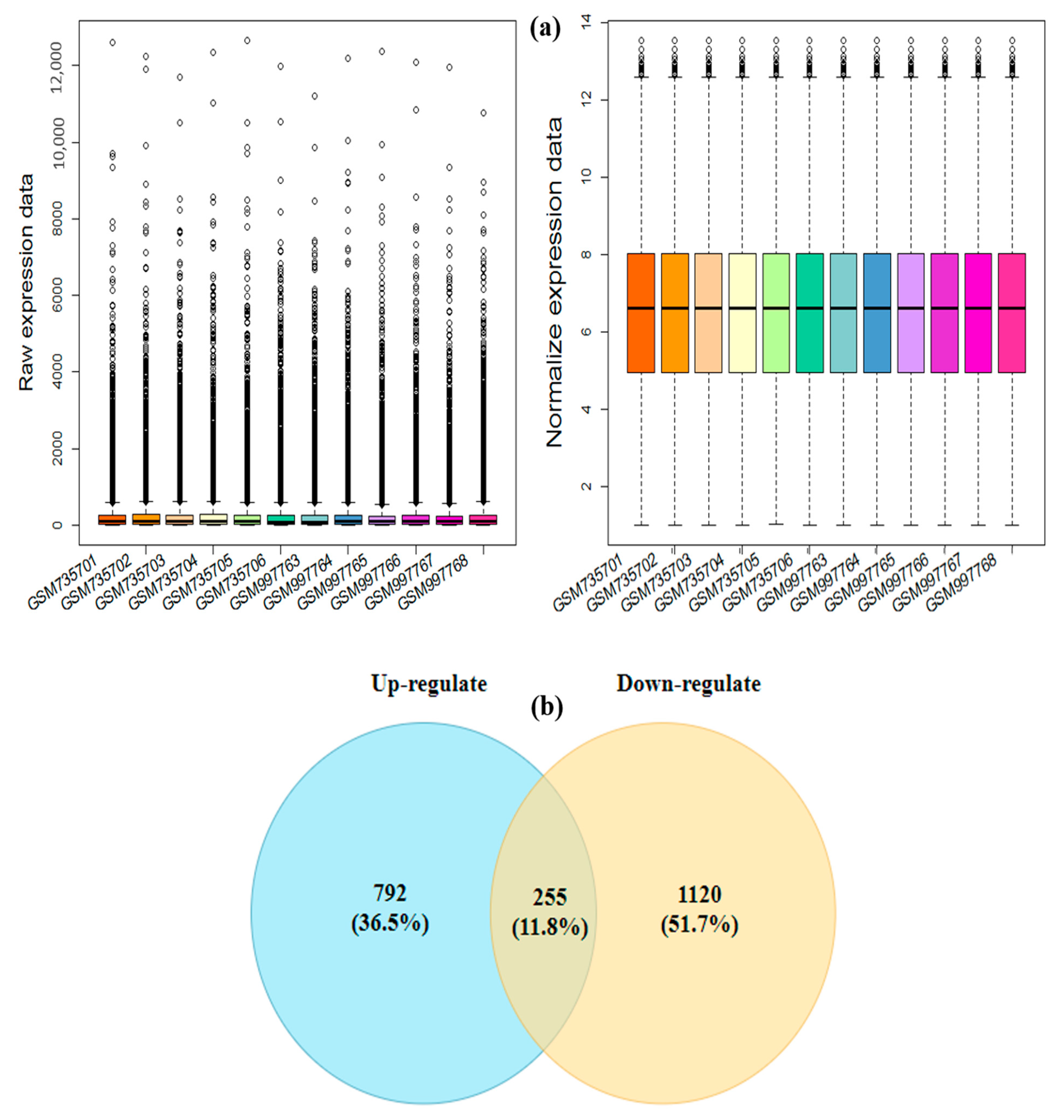
2.2. Screening and Identification of Differentially Expressed Genes
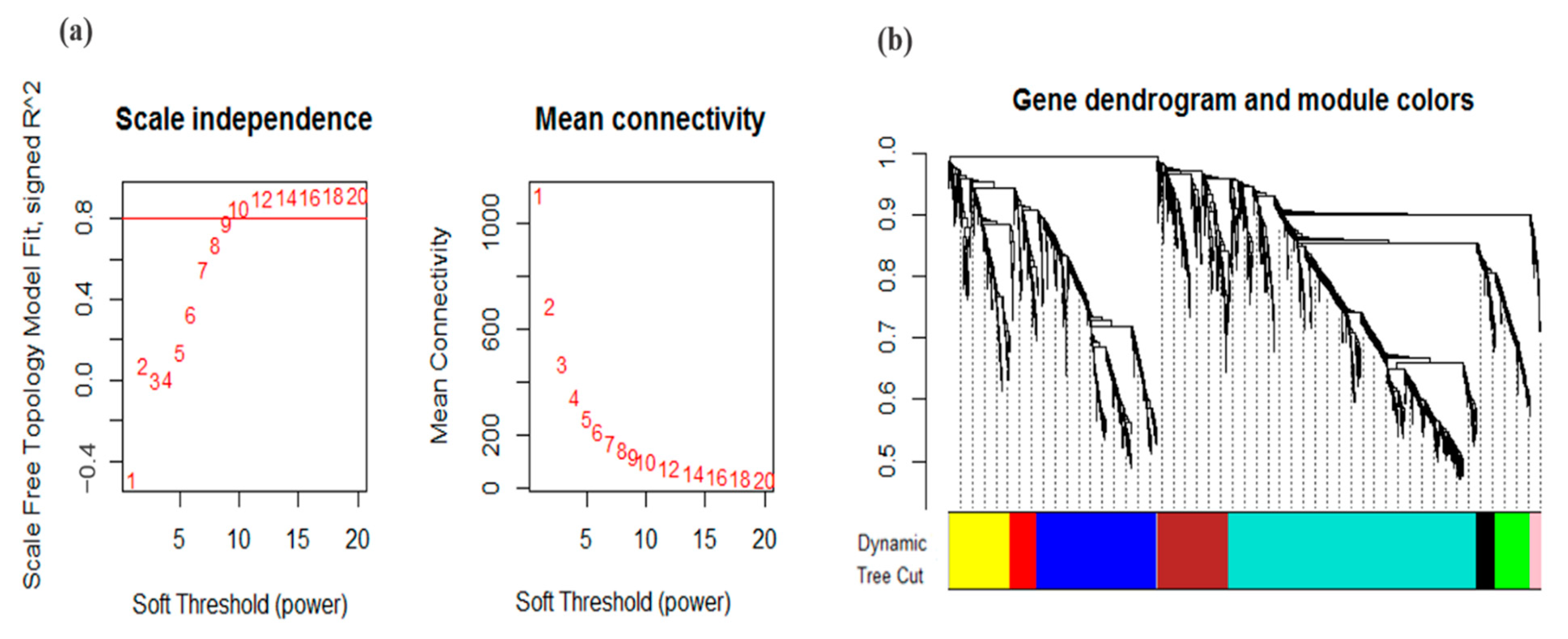
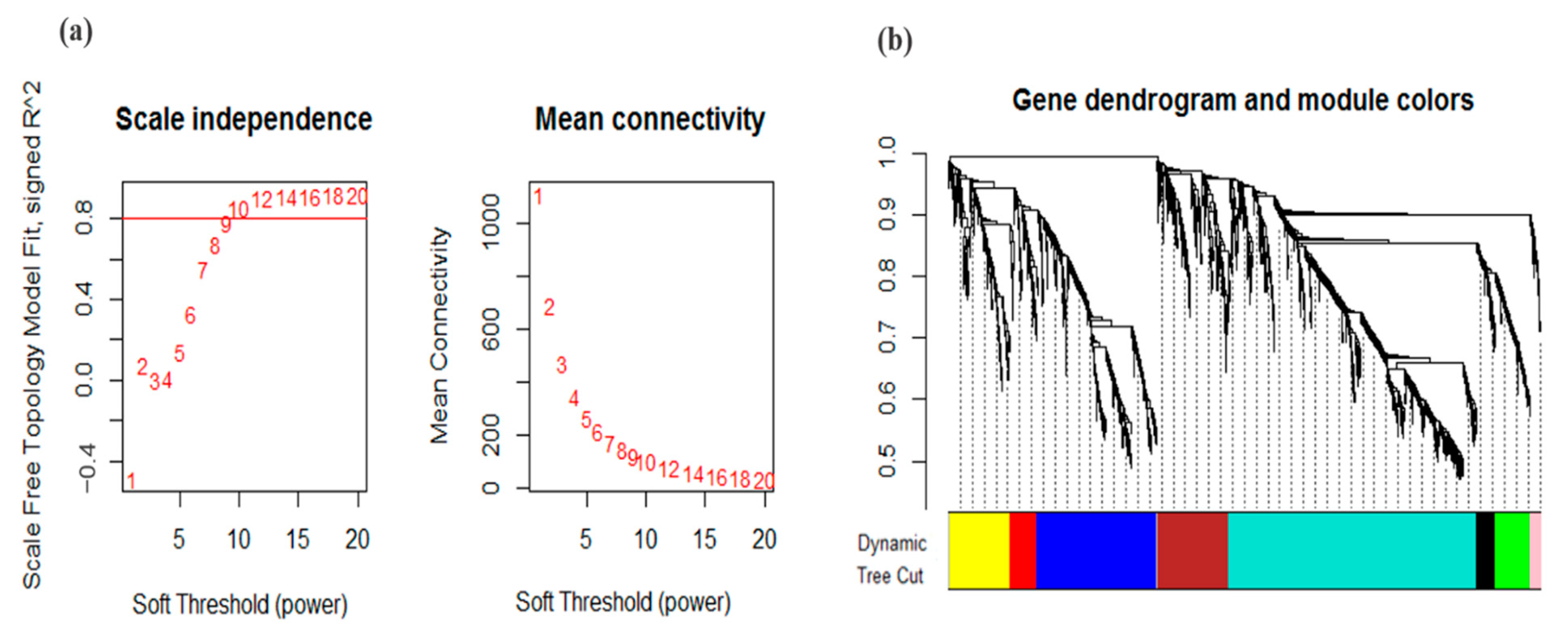
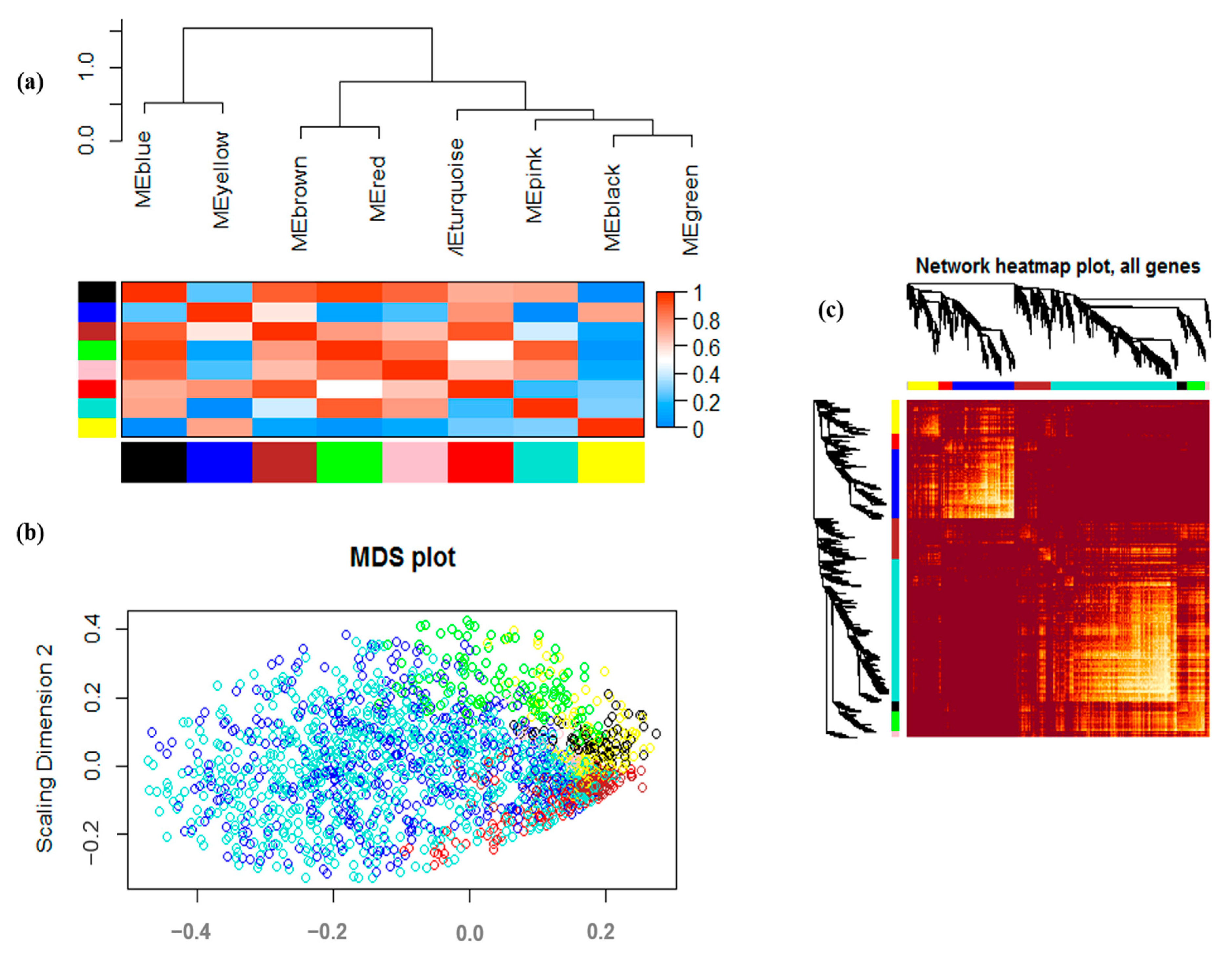
2.3. Weighted Gene Co-Expression Network Analysis
2.4. Identification of Hub Genes
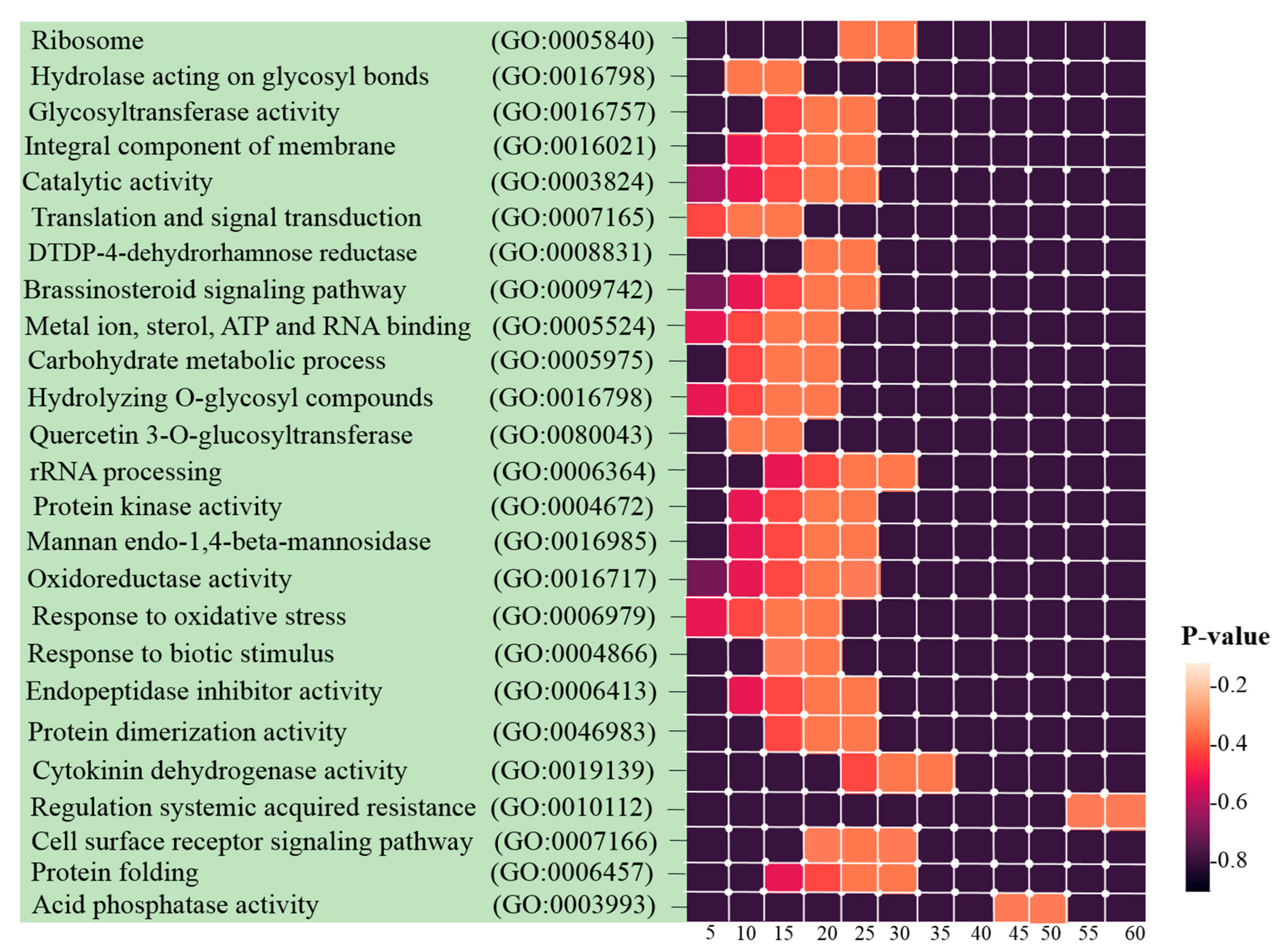
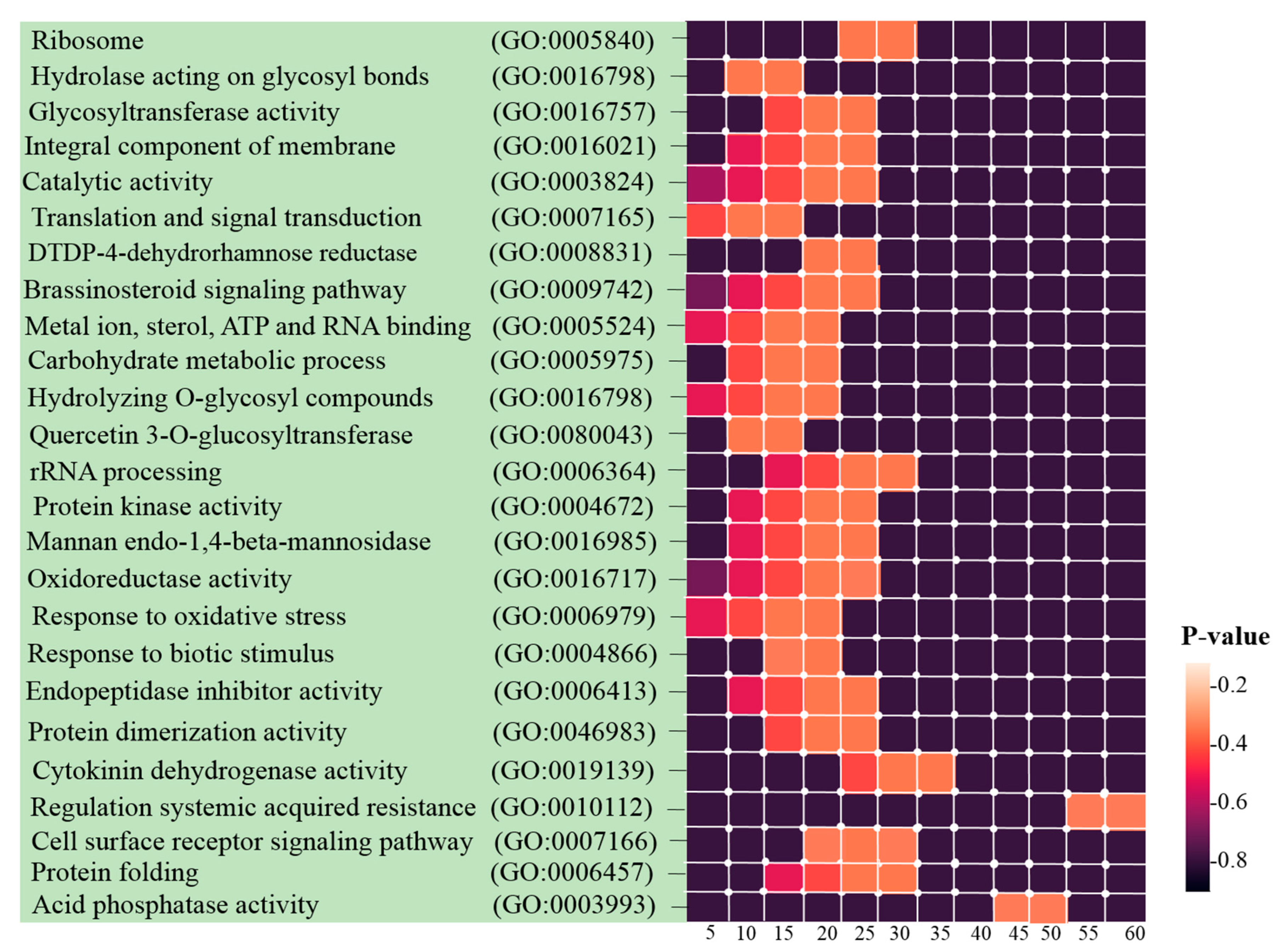
2.5. GO and Pathway Functional Enrichment Analyses
2.6. Cis-Acting Element Analysis
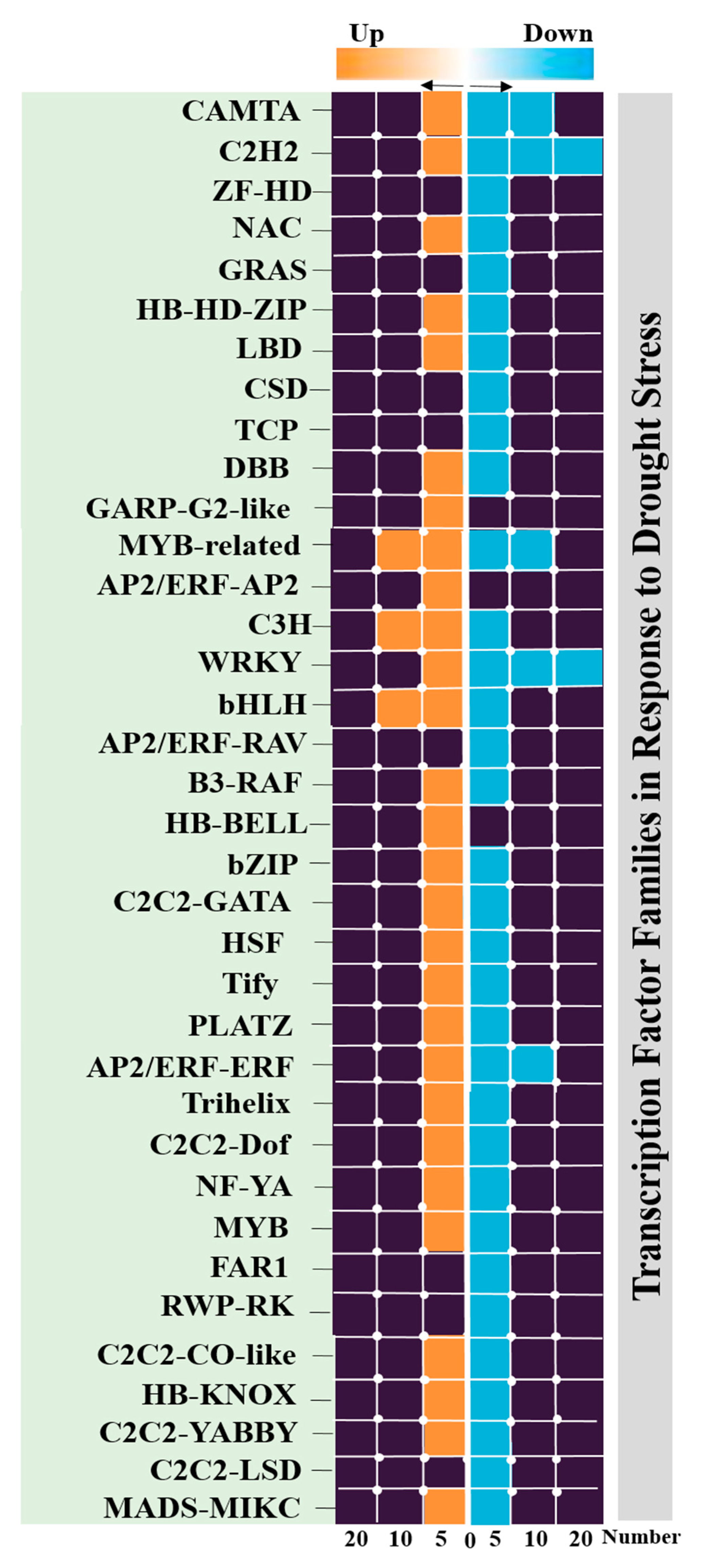
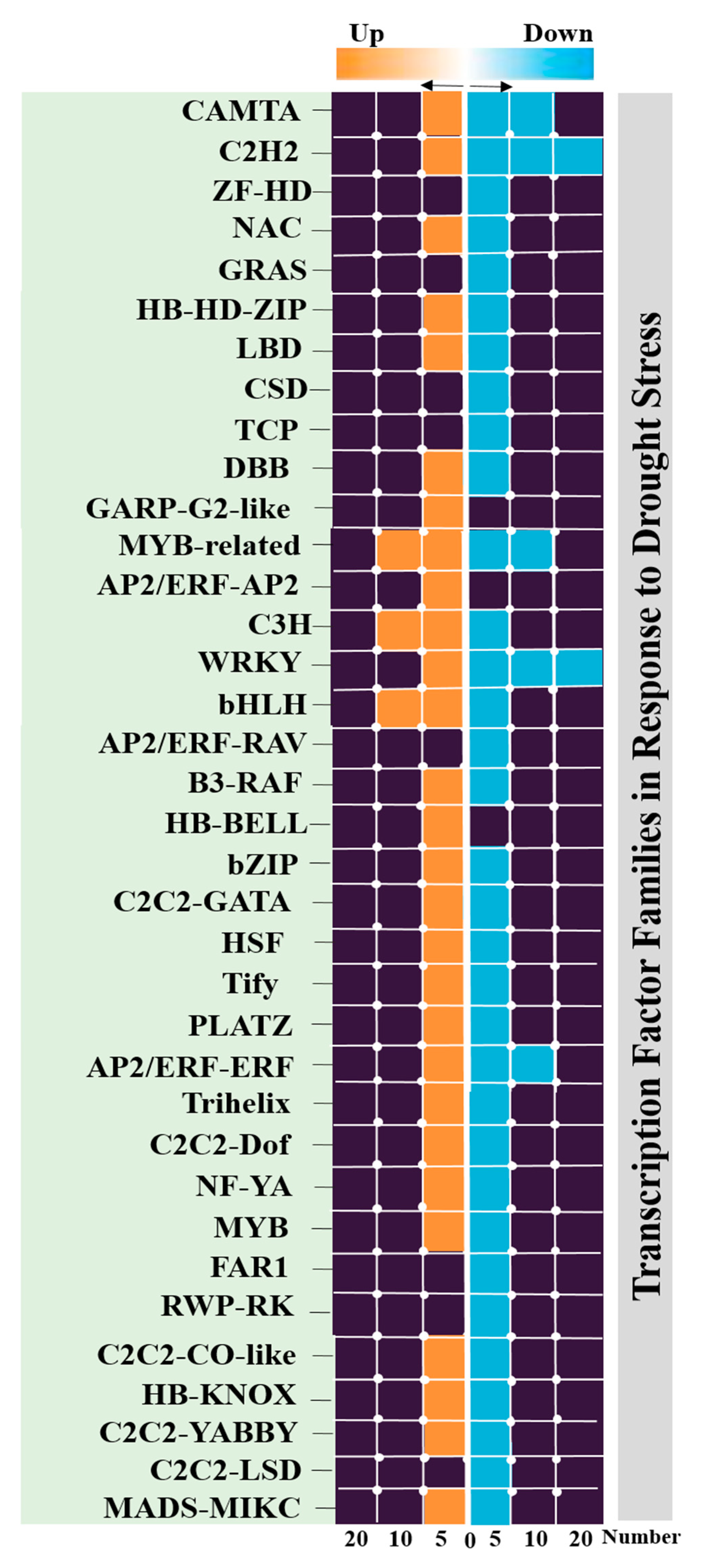
2.7. Identification of Transcription Factor Families
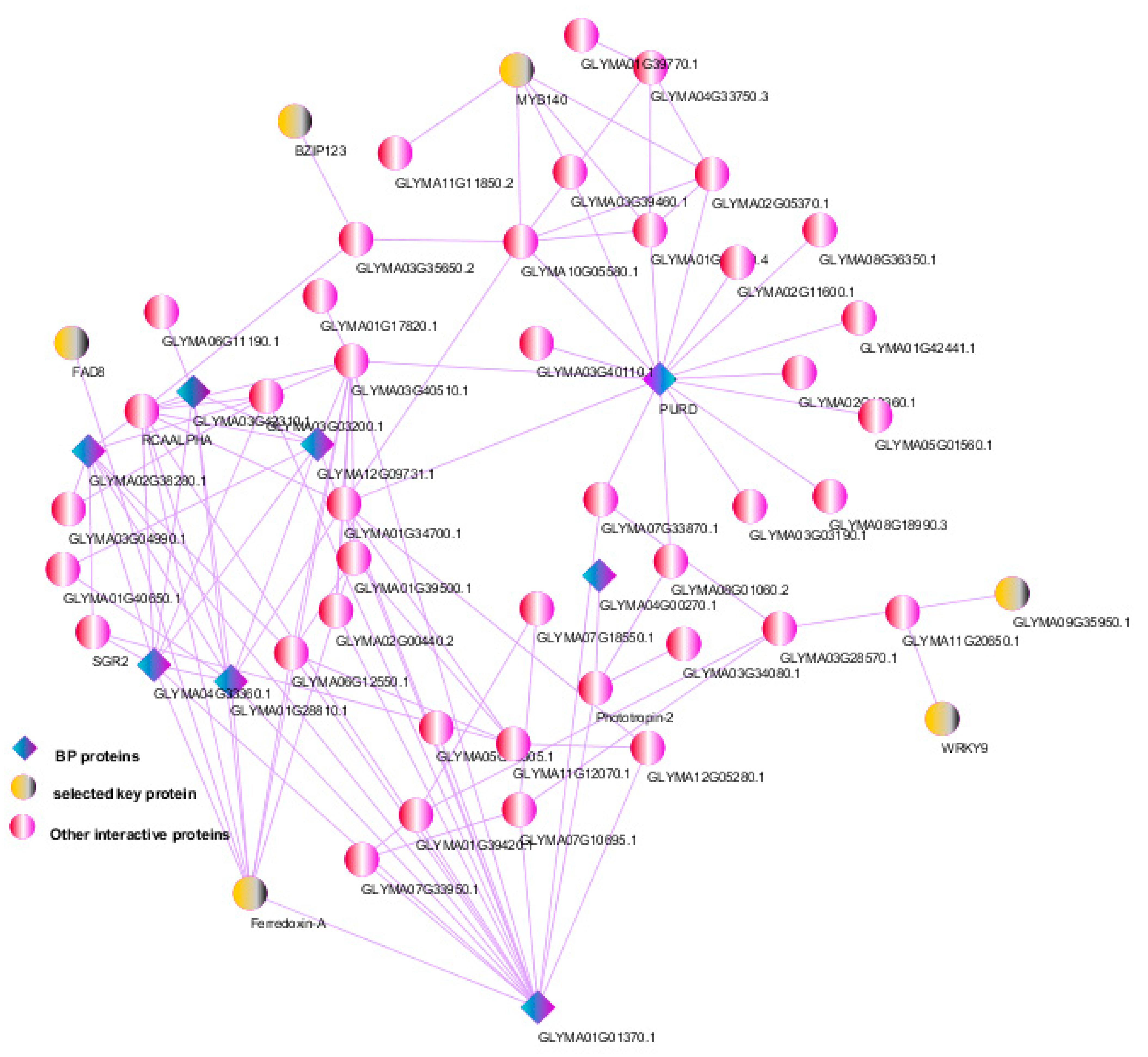
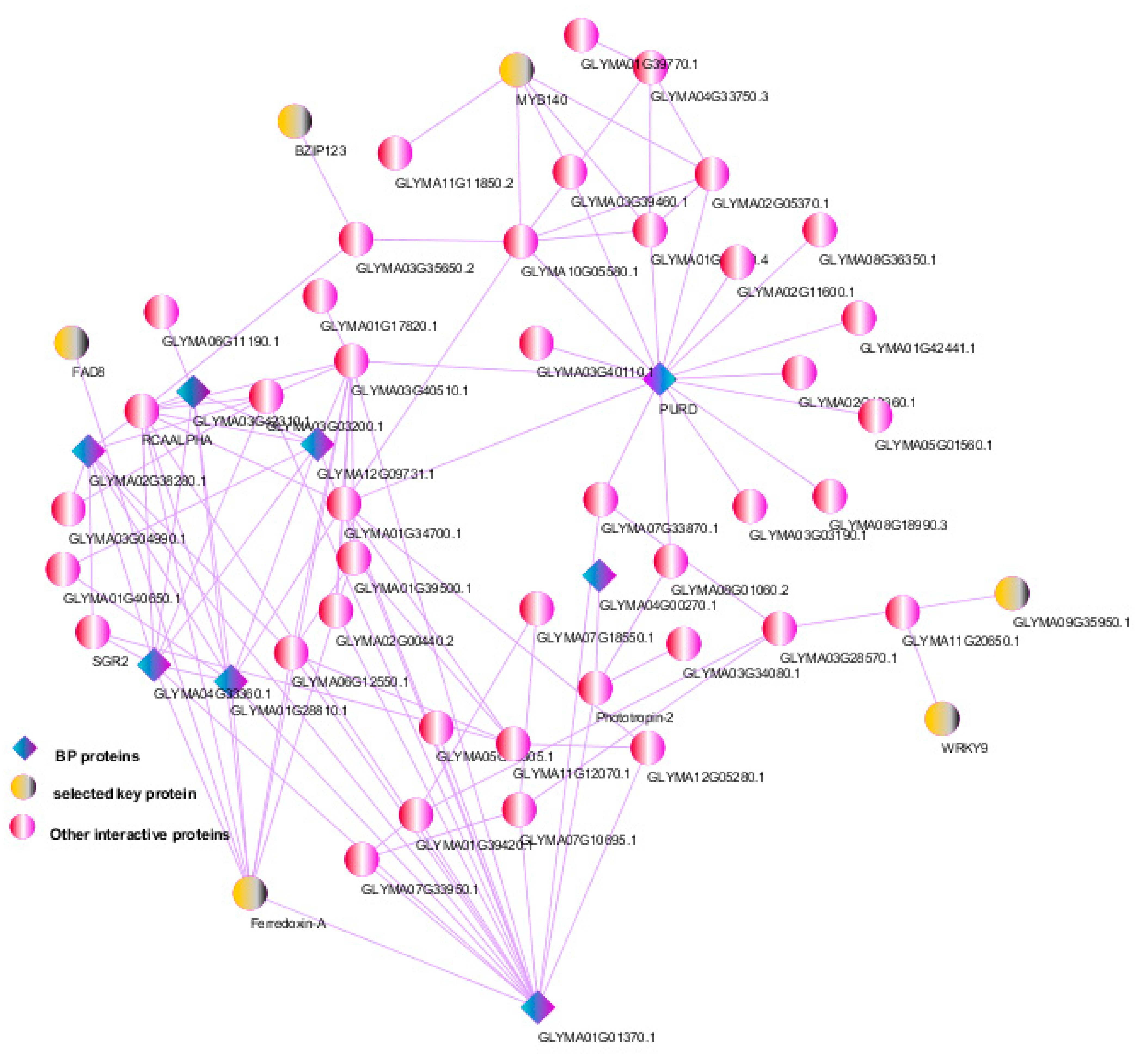
2.8. Protein–Protein Interaction (PPI) Networks
2.9. Validation Analysis
3. Results
3.1. Pre-Processing and Identification of the Drought-Responsive Core DEGs
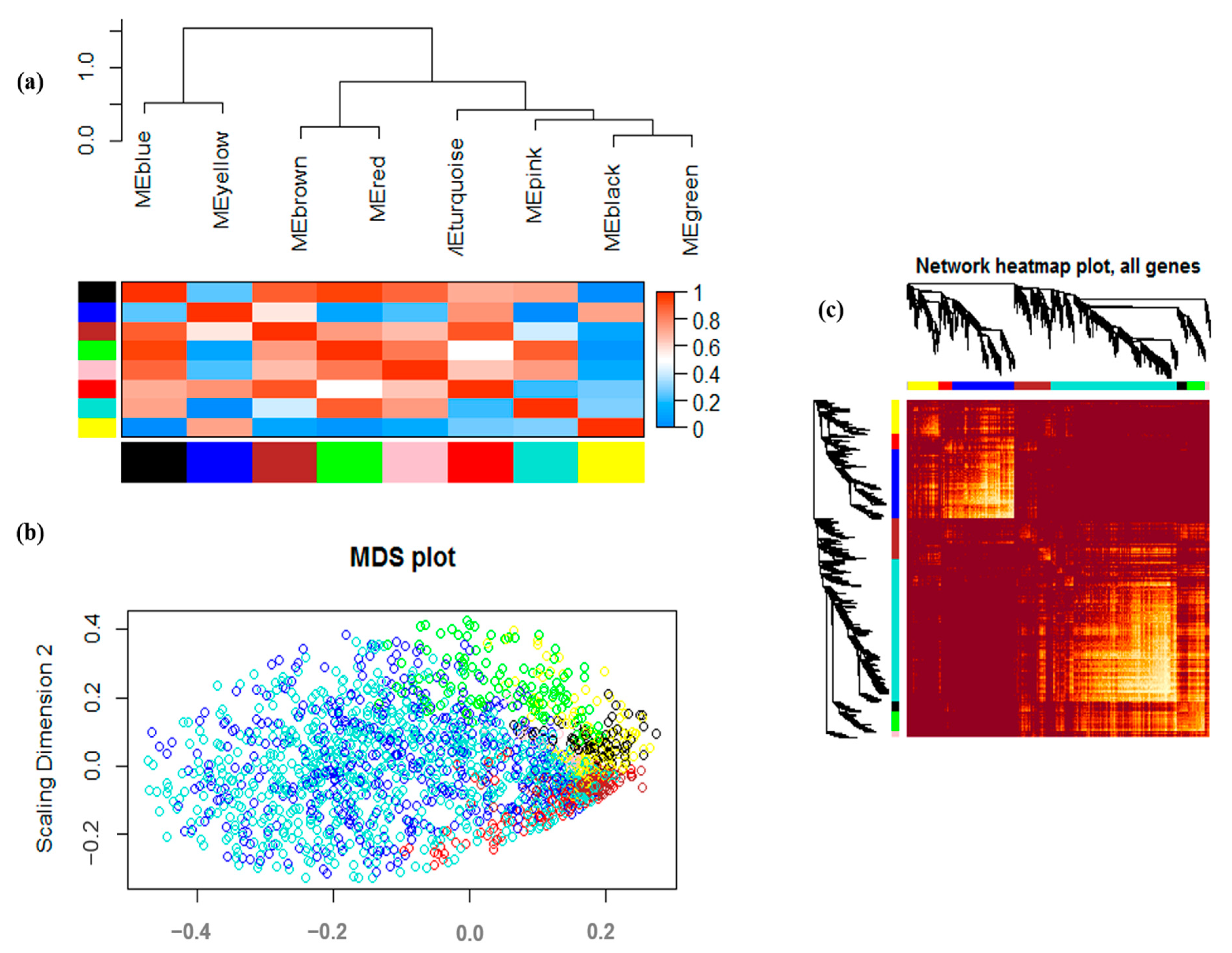
3.2. Co-Expression Analysis and Module Identification under Drought Stress
3.3. Identification of Hub Genes and Enrichment Analysis
3.4. Identification of Transcription Factors
3.5. Cis-Acting Elements Analysis and Motif Identification
3.6. Protein–Protein Interactions and Selection of Key Genes
3.7. Leave-One-Out Cross-Validation of Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prudhomme, C.; Giuntoli, I.; Robinson, E.L.; Clark, D.B.; Arnell, N.W.; Dankers, R.; Fekete, B.M.; Franssen, W.; Gerten, D.; Gosling, S.N. Hydrological droughts in the 21st century, hotspots and uncertainties from a global multimodel ensemble experiment. Proc. Natl. Acad. Sci. USA 2014, 111, 3262–3267. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Wani, S.H.; Singh, B.; Bohra, A.; Dar, Z.A.; Lone, A.A.; Pareek, A.; Singla-Pareek, S.L. Transcription factors and plants response to drought stress: Current understanding and future directions. Front. Plant Sci. 2016, 7, 1029. [Google Scholar] [CrossRef] [PubMed]
- Harb, A.; Pereira, A. Screening Arabidopsis genotypes for drought stress resistance. In Plant Reverse Genetics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 191–198. [Google Scholar]
- Pinheiro, C.; Chaves, M. Photosynthesis and drought: Can we make metabolic connections from available data? J. Exp. Bot. 2011, 62, 869–882. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wang, Z.-X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; Guo, Y. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Osakabe, K.; Shinozaki, K.; Tran, L.-S.P. Response of plants to water stress. Front. Plant Sci. 2014, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Ebeed, H.T.; El-Helely, A.A. Programmed cell death in plants: Insights into developmental and stress-induced cell death. Curr. Protein Pept. Sci. 2021, 22, 873–889. [Google Scholar] [CrossRef]
- Gong, L.; Zhang, H.; Liu, X.; Gan, X.; Nie, F.; Yang, W.; Zhang, L.; Chen, Y.; Song, Y.; Zhang, H. Ectopic expression of HaNAC1, an ATAF transcription factor from Haloxylon ammodendron, improves growth and drought tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2020, 151, 535–544. [Google Scholar] [CrossRef]
- Kumar, S.; Beena, A.; Awana, M.; Singh, A. Physiological, biochemical, epigenetic and molecular analyses of wheat (Triticum aestivum) genotypes with contrasting salt tolerance. Front. Plant Sci. 2017, 8, 1151. [Google Scholar] [CrossRef]
- Kim, S.-G.; Kim, S.-Y.; Park, C.-M. A membrane-associated NAC transcription factor regulates salt-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Planta 2007, 226, 647–654. [Google Scholar] [CrossRef]
- Ebeed, H.T. Bioinformatics Studies on the Identification of New Players and Candidate Genes to Improve Brassica Response to Abiotic Stress. In The Plant Family Brassicaceae; Springer: Berlin/Heidelberg, Germany, 2020; pp. 483–496. [Google Scholar]
- Ebeed, H.T. Omics approaches for developing abiotic stress tolerance in wheat. In Wheat Production in Changing Environments; Springer: Berlin/Heidelberg, Germany, 2019; pp. 443–463. [Google Scholar]
- Schadt, E.E.; Linderman, M.D.; Sorenson, J.; Lee, L.; Nolan, G.P. Computational solutions to large-scale data management and analysis. Nat. Rev. Genet. 2010, 11, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big data: Astronomical or genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.; Liguori, M.J. Comparison of RNA-Seq and microarray gene expression platforms for the toxicogenomic evaluation of liver from short-term rat toxicity studies. Front. Genet. 2019, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Deyholos, M.K. Making the most of drought and salinity transcriptomics. Plant Cell Environ. 2010, 33, 648–654. [Google Scholar] [CrossRef]
- Reymond, P.; Weber, H.; Damond, M.; Farmer, E.E. Differential gene expression in response to mechanical wounding and insect feeding in Arabidopsis. Plant Cell 2000, 12, 707–719. [Google Scholar] [CrossRef]
- Tahmasebi, A.; Ebrahimie, E.; Pakniyat, H.; Ebrahimi, M.; Mohammadi-Dehcheshmeh, M. Tissue-specific transcriptional biomarkers in medicinal plants: Application of large-scale meta-analysis and computational systems biology. Gene 2019, 691, 114–124. [Google Scholar] [CrossRef]
- Ebeed, H.T.; Stevenson, S.R.; Cuming, A.C.; Baker, A. Conserved and differential transcriptional responses of peroxisome associated pathways to drought, dehydration and ABA. J. Exp. Bot. 2018, 69, 4971–4985. [Google Scholar] [CrossRef]
- Sharifi, S.; Pakdel, A.; Ebrahimi, M.; Reecy, J.M.; Farsani, S.F.; Ebrahimie, E. Integration of machine learning and meta-analysis identifies the transcriptomic bio-signature of mastitis disease in cattle. PLoS ONE 2018, 13, e0191227. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Lilley, C.J.; Urwin, P.E. Identification of genes involved in the response of Arabidopsis to simultaneous biotic and abiotic stresses. Plant Physiol. 2013, 162, 2028–2041. [Google Scholar] [CrossRef] [Green Version]
- Oono, Y.; Seki, M.; Nanjo, T.; Narusaka, M.; Fujita, M.; Satoh, R.; Satou, M.; Sakurai, T.; Ishida, J.; Akiyama, K. Monitoring expression profiles of Arabidopsis gene expression during rehydration process after dehydration using ca. 7000 full-length cDNA microarray. Plant J. 2003, 34, 868–887. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Meher, P.K.; Rai, A.; Bhar, L.M.; Mandal, B.N. Statistical approaches for gene selection, hub gene identification and module interaction in gene co-expression network analysis: An application to aluminum stress in soybean (Glycine max L.). PLoS ONE 2017, 12, e0169605. [Google Scholar] [CrossRef]
- Downs, G.S.; Bi, Y.-M.; Colasanti, J.; Wu, W.; Chen, X.; Zhu, T.; Rothstein, S.J.; Lukens, L.N. A developmental transcriptional network for maize defines coexpression modules. Plant Physiol. 2013, 161, 1830–1843. [Google Scholar] [CrossRef]
- Ficklin, S.P.; Luo, F.; Feltus, F.A. The association of multiple interacting genes with specific phenotypes in rice using gene coexpression networks. Plant Physiol. 2010, 154, 13–24. [Google Scholar] [CrossRef]
- Shaik, R.; Ramakrishna, W. Genes and co-expression modules common to drought and bacterial stress responses in Arabidopsis and rice. PLoS ONE 2013, 8, e77261. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, S.; Zuo, K.; Luo, L.; Tang, K. Identification of gene modules associated with drought response in rice by network-based analysis. PLoS ONE 2012, 7, e33748. [Google Scholar] [CrossRef]
- Childs, K.L.; Davidson, R.M.; Buell, C.R. Gene coexpression network analysis as a source of functional annotation for rice genes. PLoS ONE 2011, 6, e22196. [Google Scholar] [CrossRef]
- Pereira Lima, J.J.; Buitink, J.; Lalanne, D.; Rossi, R.F.; Pelletier, S.; Da Silva, E.A.A.; Leprince, O. Molecular characterization of the acquisition of longevity during seed maturation in soybean. PLoS ONE 2017, 12, e0180282. [Google Scholar] [CrossRef] [Green Version]
- Bi, D.; Ning, H.; Liu, S.; Que, X.; Ding, K. Gene expression patterns combined with network analysis identify hub genes associated with bladder cancer. Comput. Biol. Chem. 2015, 56, 71–83. [Google Scholar] [CrossRef]
- Stumpf, M.P.; Porter, M.A. Critical truths about power laws. Science 2012, 335, 665–666. [Google Scholar] [CrossRef]
- Manavalan, L.P.; Guttikonda, S.K.; Phan Tran, L.-S.; Nguyen, H.T. Physiological and molecular approaches to improve drought resistance in soybean. Plant Cell Physiol. 2009, 50, 1260–1276. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- RC Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Farhadian, M.; Rafat, S.A.; Hasanpur, K.; Ebrahimi, M.; Ebrahimie, E. Cross-species meta-analysis of transcriptomic data in combination with supervised machine learning models identifies the common gene signature of lactation process. Front. Genet. 2018, 9, 235. [Google Scholar] [CrossRef]
- Mohammadi-Dehcheshmeh, M.; Niazi, A.; Ebrahimi, M.; Tahsili, M.; Nurollah, Z.; Ebrahimi Khaksefidi, R.; Ebrahimi, M.; Ebrahimie, E. Unified transcriptomic signature of Arbuscular mycorrhiza colonization in roots of Medicago truncatula by integration of machine learning, promoter analysis and direct merging meta-analysis. Front. Plant Sci. 2018, 9, 1550. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Soltani, Z.; Moghadam, A.; Tahmasebi, A.; Niazi, A. Integrative system biology analysis of barley transcriptome–hormonal signaling against biotic stress. bioRxiv 2021. [Google Scholar] [CrossRef]
- Balan, B.; Caruso, T.; Martinelli, F. Gaining insight into exclusive and common transcriptomic features linked with biotic stress responses in Malus. Front. Plant Sci. 2017, 8, 1569. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.11–18.13.24. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Cai, J.-H.; Tsai, J.J.; Wang, C.C. Identification of hub genes associated with development of head and neck squamous cell carcinoma by integrated bioinformatics analysis. Front. Oncol. 2020, 10, 681. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Pérez, N. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef]
- Buske, F.A.; Bodén, M.; Bauer, D.C.; Bailey, T.L. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics 2010, 26, 860–866. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Lorenzon, R.; Mariotti-Ferrandiz, E.; Aheng, C.; Ribet, C.; Toumi, F.; Pitoiset, F.; Chaara, W.; Derian, N.; Johanet, C.; Drakos, I. Clinical and multi-omics cross-phenotyping of patients with autoimmune and autoinflammatory diseases: The observational TRANSIMMUNOM protocol. BMJ Open 2018, 8, e021037. [Google Scholar] [CrossRef]
- Tahmasebi, A.; Ashrafi-Dehkordi, E.; Shahriari, A.G.; Mazloomi, S.M.; Ebrahimie, E. Integrative meta-analysis of transcriptomic responses to abiotic stress in cotton. Prog. Biophys. Mol. Biol. 2019, 146, 112–122. [Google Scholar] [CrossRef]
- Li, A.; Horvath, S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics 2007, 23, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Silva, F.; Moharana, K.C.; Machado, F.B.; Venancio, T.M. Exploring the complexity of soybean (Glycine max) transcriptional regulation using global gene co-expression networks. Planta 2020, 252, 104. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Kühn, C.; Tegeder, M.; Frommer, W.B. Sucrose transport in higher plants. Int. Rev. Cytol. 1997, 178, 41–71. [Google Scholar]
- Chen, F.; Dixon, R.A. Lignin modification improves fermentable sugar yields for biofuel production. Nat. Biotechnol. 2007, 25, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.; Kristensen, J.B.; Felby, C. Enzymatic conversion of lignocellulose into fermentable sugars: Challenges and opportunities. Biofuels Bioprod. Biorefining 2007, 1, 119–134. [Google Scholar] [CrossRef]
- Yan, J.; Aznar, A.; Chalvin, C.; Birdseye, D.S.; Baidoo, E.E.; Eudes, A.; Shih, P.M.; Loqué, D.; Zhang, A.; Scheller, H.V. Increased drought tolerance in plants engineered for low lignin and low xylan content. Biotechnol. Biofuels 2018, 11, 195. [Google Scholar] [CrossRef]
- Agarwal, P.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol. Plant. 2010, 54, 201–212. [Google Scholar] [CrossRef]
- Sharma, R.; Singh, G.; Bhattacharya, S.; Singh, A. Comparative transcriptome meta-analysis of Arabidopsis thaliana under drought and cold stress. PLoS ONE 2018, 13, e0203266. [Google Scholar]
- Jaleel, C.A.; Manivannan, P.; Wahid, A.; Farooq, M.; Al-Juburi, H.J.; Somasundaram, R.; Panneerselvam, R. Drought stress in plants: A review on morphological characteristics and pigments composition. Int. J. Agric. Biol. 2009, 11, 100–105. [Google Scholar]
- Liu, S.; Zenda, T.; Dong, A.; Yang, Y.; Wang, N.; Duan, H. Global transcriptome and weighted gene co-expression network analyses of growth-stage-specific drought stress responses in maize. Front. Genet. 2021, 12, 645443. [Google Scholar] [CrossRef]
- Divya Bhanu, B.; Ulaganathan, K.; Shanker, A.K.; Desai, S. RNA-seq analysis of irrigated vs. water stressed transcriptomes of Zea mays cultivar Z59. Front. Plant Sci. 2016, 7, 239. [Google Scholar] [CrossRef] [PubMed]
- Aranjuelo, I.; Molero, G.; Erice, G.; Avice, J.C.; Nogués, S. Plant physiology and proteomics reveals the leaf response to drought in alfalfa (Medicago sativa L.). J. Exp. Bot. 2011, 62, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, C.; Chen, H.Y.; Ruan, H. Response of plants to water stress: A meta-analysis. Front. Plant Sci. 2020, 11, 978. [Google Scholar] [CrossRef]
- Kron, A.P.; Souza, G.M.; Ribeiro, R.V. Water deficiency at different developmental stages of Glycine max can improve drought tolerance. Bragantia 2008, 67, 43–49. [Google Scholar] [CrossRef]
- Bouchemal, K.; Bouldjadj, R.; Belbekri, M.N.; Ykhlef, N.; Djekoun, A. Differences in antioxidant enzyme activities and oxidative markers in ten wheat (Triticum durum Desf.) genotypes in response to drought, heat and paraquat stress. Arch. Agron. Soil Sci. 2017, 63, 710–722. [Google Scholar] [CrossRef]
- Anjum, N.A.; Ahmad, I.; Mohmood, I.; Pacheco, M.; Duarte, A.C.; Pereira, E.; Umar, S.; Ahmad, A.; Khan, N.A.; Iqbal, M. Modulation of glutathione and its related enzymes in plants’ responses to toxic metals and metalloids—A review. Environ. Exp. Bot. 2012, 75, 307–324. [Google Scholar] [CrossRef]
- Rontein, D.; Basset, G.; Hanson, A.D. Metabolic engineering of osmoprotectant accumulation in plants. Metab. Eng. 2002, 4, 49–56. [Google Scholar] [CrossRef]
- Ozturk, M.; Turkyilmaz Unal, B.; García-Caparrós, P.; Khursheed, A.; Gul, A.; Hasanuzzaman, M. Osmoregulation and its actions during the drought stress in plants. Physiol. Plant. 2021, 172, 1321–1335. [Google Scholar] [CrossRef]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef]
- Reguera, M.; Peleg, Z.; Abdel-Tawab, Y.M.; Tumimbang, E.B.; Delatorre, C.A.; Blumwald, E. Stress-induced cytokinin synthesis increases drought tolerance through the coordinated regulation of carbon and nitrogen assimilation in rice. Plant Physiol. 2013, 163, 1609–1622. [Google Scholar] [CrossRef]
- Chen, J.B.; Yang, J.W.; Zhang, Z.Y.; Feng, X.F.; Wang, S.M. Two P5CS genes from common bean exhibiting different tolerance to salt stress in transgenic Arabidopsis. J. Genet. 2013, 92, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Kasirajan, L.; Hoang, N.V.; Furtado, A.; Botha, F.C.; Henry, R.J. Transcriptome analysis highlights key differentially expressed genes involved in cellulose and lignin biosynthesis of sugarcane genotypes varying in fiber content. Sci. Rep. 2018, 8, 11612. [Google Scholar] [CrossRef]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genom. 2012, 13, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Shin, R.; Burch, A.Y.; Huppert, K.A.; Tiwari, S.B.; Murphy, A.S.; Guilfoyle, T.J.; Schachtman, D.P. The Arabidopsis transcription factor MYB77 modulates auxin signal transduction. Plant Cell 2007, 19, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xing, L.; Wang, X.; Hou, Y.-J.; Gao, J.; Wang, P.; Duan, C.-G.; Zhu, X.; Zhu, J.-K. The ABA receptor PYL8 promotes lateral root growth by enhancing MYB77-dependent transcription of auxin-responsive genes. Sci. Signal. 2014, 7, ra53. [Google Scholar] [CrossRef]
- Zhang, D.; Tong, J.; Xu, Z.; Wei, P.; Xu, L.; Wan, Q.; Huang, Y.; He, X.; Yang, J.; Shao, H. Soybean C2H2-type zinc finger protein GmZFP3 with conserved QALGGH motif negatively regulates drought responses in transgenic Arabidopsis. Front. Plant Sci. 2016, 7, 325. [Google Scholar] [CrossRef]
- Rojo, E.; Solano, R.; Sánchez-Serrano, J.J. Interactions between signaling compounds involved in plant defense. J. Plant Growth Regul. 2003, 22, 82–98. [Google Scholar] [CrossRef]
- Lemon, B.; Tjian, R. Orchestrated response: A symphony of transcription factors for gene control. Genes Dev. 2000, 14, 2551–2569. [Google Scholar] [CrossRef]
- Sandelin, A.; Alkema, W.; Engström, P.; Wasserman, W.W.; Lenhard, B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004, 32, D91–D94. [Google Scholar] [CrossRef]
- Ruta, V.; Longo, C.; Lepri, A.; De Angelis, V.; Occhigrossi, S.; Costantino, P.; Vittorioso, P. The DOF transcription factors in seed and seedling development. Plants 2020, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Klees, S.; Lange, T.M.; Bertram, H.; Rajavel, A.; Schlüter, J.-S.; Lu, K.; Schmitt, A.O.; Gültas, M. In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data. Int. J. Mol. Sci. 2021, 22, 789. [Google Scholar] [CrossRef]
- Guerriero, G.; Piasecki, E.; Berni, R.; Xu, X.; Legay, S.; Hausman, J.-F. Identification of callose synthases in stinging nettle and analysis of their expression in different tissues. Int. J. Mol. Sci. 2020, 21, 3853. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Xu, L.; Dossa, K.; Zhou, K.; Zhu, M.; Xie, H.; Tang, S.; Yu, Y.; Guo, X.; Zhou, B. Identification of putative drought-responsive genes in rice using gene co-expression analysis. Bioinformation 2019, 15, 480. [Google Scholar] [CrossRef]
- Davoudi, M.; Song, M.; Zhang, M.; Chen, J.; Lou, Q. Long-distance control of the scion by the rootstock under drought stress as revealed by transcriptome sequencing and mobile mRNA identification. Hortic. Res. 2022, 9, uhab033. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Raina, S.K.; Kumar, M.; Aher, L.; Ratnaparkhe, M.B.; Rane, J.; Kachroo, A. Modulation of GmFAD3 Expression Alters Abiotic Stress Responses in Soybean. Plant Mol. Biol. 2022, 110, 199–218. [Google Scholar] [CrossRef]
- Sui, N.; Wang, Y.; Liu, S.; Yang, Z.; Wang, F.; Wan, S. Transcriptomic and physiological evidence for the relationship between unsaturated fatty acid and salt stress in peanut. Front. Plant Sci. 2018, 9, 7. [Google Scholar] [CrossRef]
- Zhang, M.; Barg, R.; Yin, M.; Gueta-Dahan, Y.; Leikin-Frenkel, A.; Salts, Y.; Shabtai, S.; Ben-Hayyim, G. Modulated fatty acid desaturation via overexpression of two distinct ω-3 desaturases differentially alters tolerance to various abiotic stresses in transgenic tobacco cells and plants. Plant J. 2005, 44, 361–371. [Google Scholar] [CrossRef]
- Huang, H.-E.; Ho, M.-H.; Chang, H.; Chao, H.-Y.; Ger, M.-J. Overexpression of plant ferredoxin-like protein promotes salinity tolerance in rice (Oryza sativa). Plant Physiol. Biochem. 2020, 155, 136–146. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Huang, L.-F.; Hase, T.; Huang, H.-E.; Feng, T.-Y. Expression of plant ferredoxin-like protein (PFLP) enhances tolerance to heat stress in Arabidopsis thaliana. New Biotechnol. 2015, 32, 235–242. [Google Scholar] [CrossRef]
- He, L.; Li, M.; Qiu, Z.; Chen, D.; Zhang, G.; Wang, X.; Chen, G.; Hu, J.; Gao, Z.; Dong, G. Primary leaf-type ferredoxin 1 participates in photosynthetic electron transport and carbon assimilation in rice. Plant J. 2020, 104, 44–58. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Type | Platform | Control | Treatment | Tissue | Released |
|---|---|---|---|---|---|---|
| E-GEOD-40627 | Response to drought | GPL13674, Affymetrix | 3 | 3 | Leaf | 10 November 2012 |
| E-GEOD-65553 | Response to drought | GPL13674, Affymetrix | 9 | 9 | Root | 2 July 2015 |
| E-GEOD-29663 | Response to drought | GPL13674, Affymetrix | 3 | 3 | Leaf | 2 April 2012 |
| Total samples | 30 |
| Module Name | Number of Genes |
|---|---|
| Blue | 441 |
| Brown | 258 |
| Green | 128 |
| Red | 99 |
| Turquois | 911 |
| Yellow | 220 |
| Black | 68 |
| Pink | 36 |
| Motif Name | Motif Logo | E-Value | Width | Best Match in JASPAR and PLACE | Significant GO Terms Identified by GOMO |
|---|---|---|---|---|---|
| Motif 1 |  | 8.0 × 10−100 | 41 | MA1403.1 (BBR/BBC) | MF: transcription factor activity CC: nucleus CC: plasma membrane BP: regulation of transcription, DNA-dependent BP: protein amino acid phosphorylation |
| Motif 2 |  | 1.1 × 10−44 | 20 | MA1268.1 (Dof) | MF: transcription factor activity CC: nucleus MF: protein serine/threonine kinase activity BP: protein amino acid phosphorylation BP: regulation of transcription, DNA-dependent |
| Motif 3 | 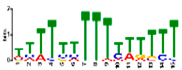 | 9.6 × 10−27 | 15 | MA1268.1 (Dof) | MF: transcription factor activity BP: regulation of transcription CC: plasma membrane CC: nucleus BP: response to water deprivation |
| Motif 4 | 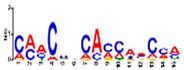 | 1.6 × 10−17 | 15 | MA1890.1 (C2H2) | CC: chloroplast MF: transcription factor activity BP: photosynthetic electron transport in photosystem I |
| Motif 5 | 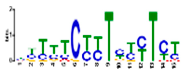 | 1.7 × 10−5 | 15 | MA1403.1 (BBR/BBC) | MF: transcription factor activity CC: nucleus CC: plasma membrane BP: regulation of transcription, DNA-dependent MF: protein binding |
| Motif 6 |  | 1.4 × 10−10 | 21 | MA1281.1 (Dof) | MF: transcription factor activity CC: plasma membrane CC: nucleus BP: regulation of transcription BP: response to water deprivation |
| Motif 7 | 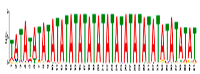 | 5.3 × 10−5 | 41 | MA0386.1 (Fox) | MF: transcription factor activity CC: endomembrane system BP: regulation of transcription, DNA-dependent BP: oligopeptide transport MF: protein heterodimerization activity |
| Motif 8 |  | 9.1 × 10−1 | 29 | MA1354.1 (MYB-related) | CC: chloroplast |
| Motif 9 | 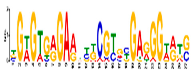 | 1.9 × 10−3 | 27 | MA1892.1 (C2H2) | CC: chloroplast MF: transcription factor activity CC: nucleus BP: DNA replication initiation BP: developmental growth |
| Motif 10 |  | 1.1 × 10−4 | 21 | MA1723.1 (C2H2) | MF: transcription factor activity CC: nucleus BP: regulation of transcription, DNA-dependent CC: plasma membrane MF: protein binding |
| Motif 11 |  | 2.1 × 10−4 | 21 | MA1766.1 (MYB) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahriari, A.G.; Soltani, Z.; Tahmasebi, A.; Poczai, P. Integrative System Biology Analysis of Transcriptomic Responses to Drought Stress in Soybean (Glycine max L.). Genes 2022, 13, 1732. https://doi.org/10.3390/genes13101732
Shahriari AG, Soltani Z, Tahmasebi A, Poczai P. Integrative System Biology Analysis of Transcriptomic Responses to Drought Stress in Soybean (Glycine max L.). Genes. 2022; 13(10):1732. https://doi.org/10.3390/genes13101732
Chicago/Turabian StyleShahriari, Amir Ghaffar, Zahra Soltani, Aminallah Tahmasebi, and Péter Poczai. 2022. "Integrative System Biology Analysis of Transcriptomic Responses to Drought Stress in Soybean (Glycine max L.)" Genes 13, no. 10: 1732. https://doi.org/10.3390/genes13101732
APA StyleShahriari, A. G., Soltani, Z., Tahmasebi, A., & Poczai, P. (2022). Integrative System Biology Analysis of Transcriptomic Responses to Drought Stress in Soybean (Glycine max L.). Genes, 13(10), 1732. https://doi.org/10.3390/genes13101732