
Exome Sequencing Identifies a Biallelic GALNS Variant (p.Asp233Asn) Causing Mucopolysaccharidosis Type IVA in a Pakistani Consanguineous Family

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Ethical Approval and Patient Consent
2.2. Sample Collection and DNA Extraction
2.3. Whole Exome Sequencing
2.4. Data Analysis and Filtration
2.5. Sanger Sequencing
3. Results
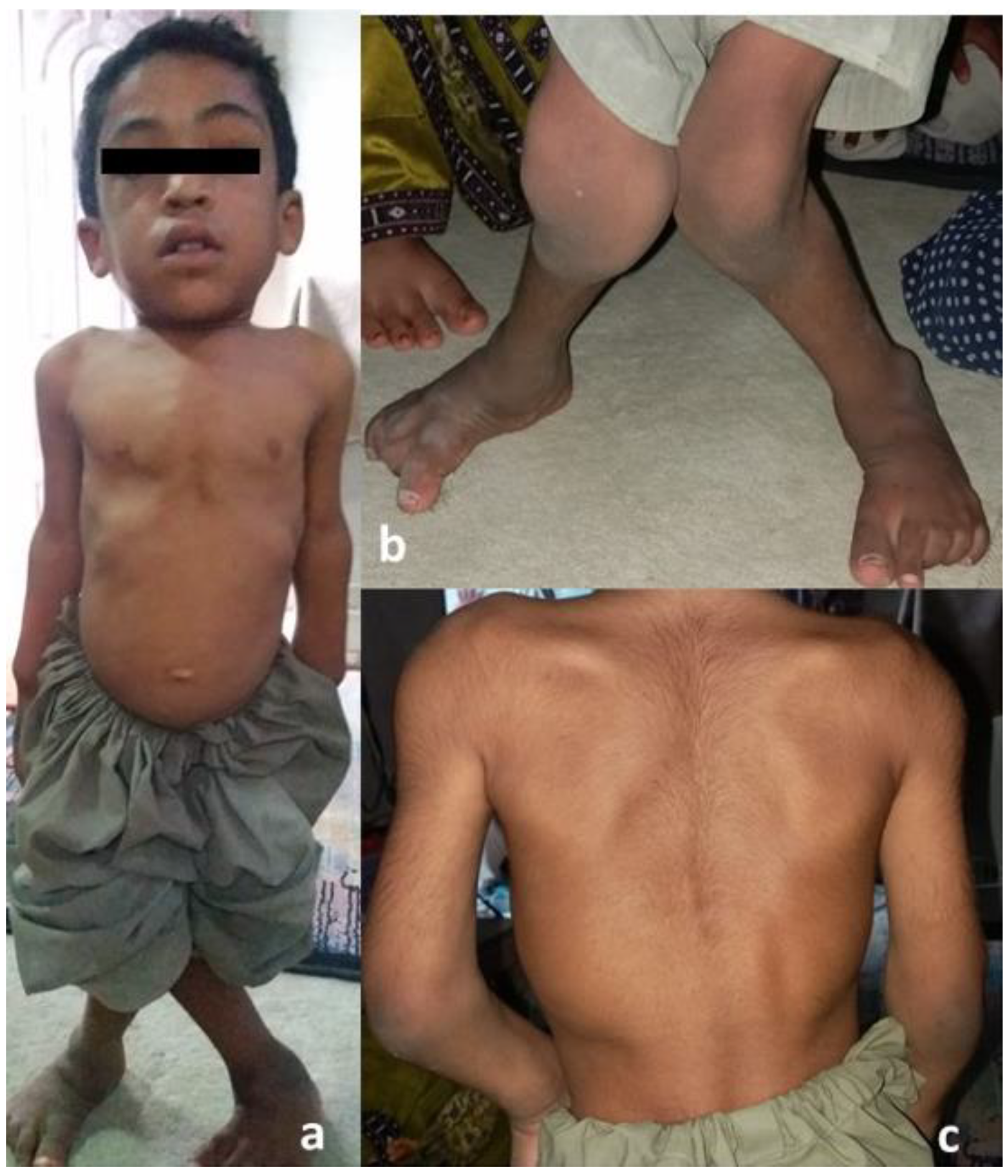
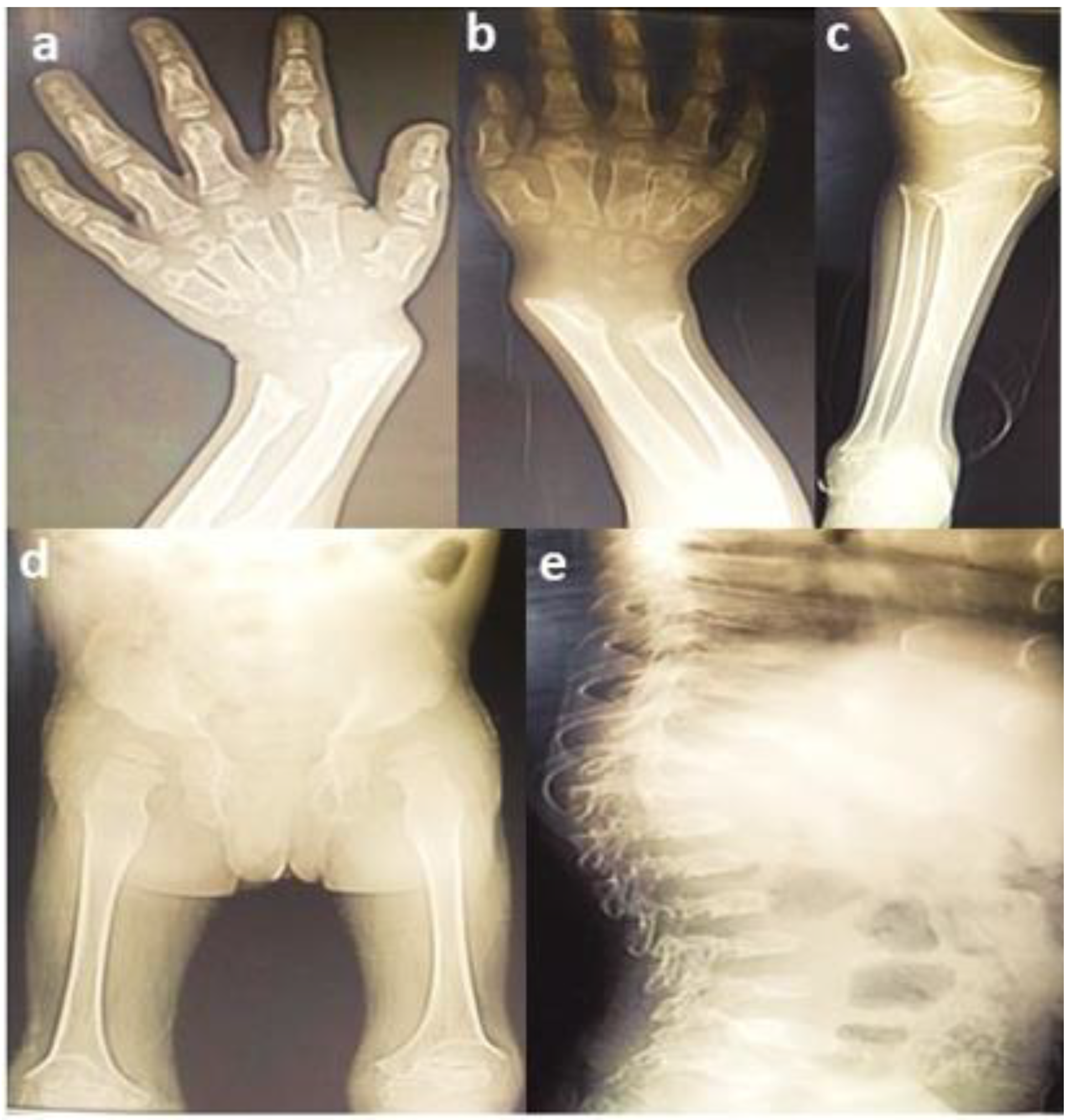
3.1. Clinical Findings
3.2. Genetic Findings
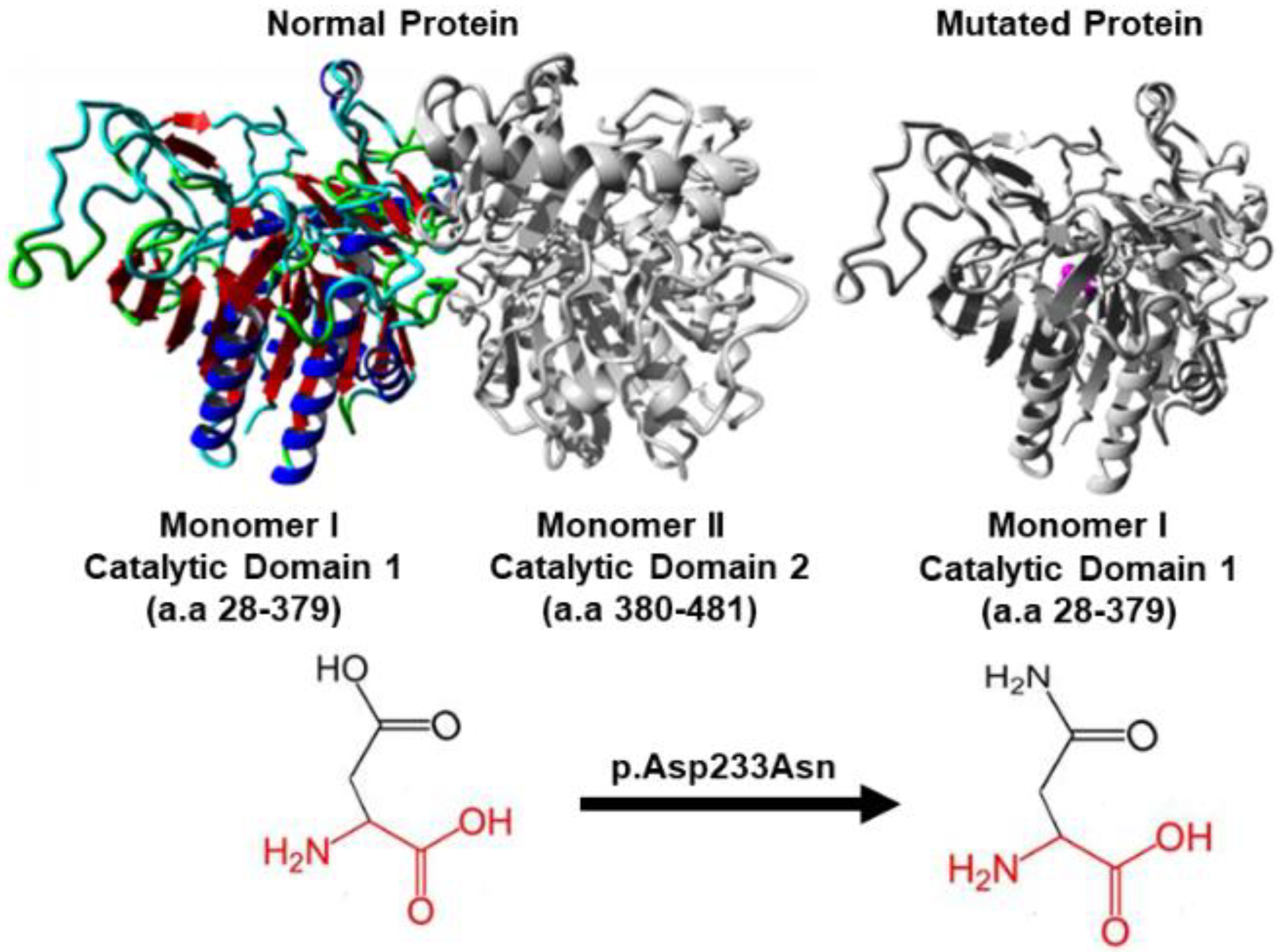
3.3. In Silico Protein Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hao, T.K.; Chi, N.T.D.; Duc, N.T.H.; Hoa, N.T.K. A case study of three patients with mucopolysaccharidoses in Hue Central Hospital. SAGE Open Med. Case Rep. 2020, 8, 2050313X2093824. [Google Scholar]
- Ramalingam, S.M.; Srinivasan, D.; ArunKumar, S.; ChiriyanKandath, J.L.; Kaliamoorthy, S. Morquio’s Syndrome: A Case Report of Two Siblings. Case Rep. Dent. 2017, 2017, 6176372. [Google Scholar]
- Sahin, K.; Elevli, M.; Kalkan, T. Mucopolysaccharidosis type 4 (Morquio syndrome): A case report. SiSli Etfal Hastan. Tip Bul./Med. Bull. Sisli Hosp. 2017, 3, 243–246. [Google Scholar] [CrossRef]
- Harmatz, P.; Mengel, K.E.; Giugliani, R.; Valayannopoulos, V.; Lin, S.-P.; Parini, R.; Guffon, N.; Burton, B.K.; Hendriksz, C.J.; Mitchell, J.; et al. The Morquio A Clinical Assessment Program: Baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol. Genet. Metab. 2013, 109, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Regier, D.S.; Oetgen, M.; Tanpaiboon, P. Mucopolysaccharidosis Type IVA Synonyms: Morquio A Disease, Morquio Syndrome Type A, MPS IVA. Available online: https://www.ncbi.nlm.nih.gov/books/NBK148668/ (accessed on 10 December 2021).
- Morishita, K.; Petty, R.E. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology 2011, 50, v19–v25. [Google Scholar] [CrossRef] [PubMed]
- White, K.K. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology 2011, 50, v26–v33. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; Volumes 1–3. [Google Scholar]
- Hamosh, A.; Sobreira, N.; Hoover-Fong, J.; Sutton, V.R.; Boehm, C.; Schiettecatte, F.; Valle, D. PhenoDB: A New Web-Based Tool for the Collection, Storage, and Analysis of Phenotypic Features. Hum. Mutat. 2013, 34, 566–571. [Google Scholar] [CrossRef]
- Rodrigues, E.d.S.; Griffith, S.; Martin, R.; Antonescu, C.; Posey, J.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Doheny, K.F.; Lupski, J.R.; Valle, D.; et al. Variant-level matching for diagnosis and discovery: Challenges and opportunities. Hum. Mutat. 2022, 43, 782–790. [Google Scholar] [CrossRef]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022, 43, 1012–1030. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Venselaar, H.; Beek, T.A.H.T.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sengar, K.; Subramanian, A.; Satyarthee, G. Morquio Syndrome Presenting with Dural Band Pathology: A Case Report. J. Lab. Physicians 2020, 12, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Cheema, H.A.; Malik, H.S.; Hashmi, M.A.; Fayyaz, Z.; Mushtaq, I.; Shahzadi, N. Mucopolysaccharidoses—Clinical Spectrum and Frequency of Different Types. J. Coll. Physicians Surg. Pak. 2017, 27, 80–83. [Google Scholar] [PubMed]
- Ullah, I. In silico analysis of GALNS mutations identified in fifty six Pakistani individuals with Morquio A syndrome. In Proceedings of the National Biology Conference (Researchgate, 2017), Kohat, Pakistan, 26–27 April 2017. [Google Scholar]
- Bidchol, A.M.; Dalal, A.; Shah, H.; Nampoothiri, S.; Kabra, M.; Gupta, N.; Danda, S.; Gowrishankar, K.; Phadke, S.R.; Kapoor, S.; et al. GALNS mutations in Indian patients with mucopolysaccharidosis IVA. Am. J. Med. Genet. Part A 2014, 164, 2793–2801. [Google Scholar] [CrossRef]
- Ge, Z.; Mao, J.; Shen, H.; Xu, Y.; Fu, H.; Zhang, W.; Li, D. Clinical and genetic characteristics of concomitant Mucopolysaccharidosis type IVA and neurogenic bladder in children: Two case reports and literature review. BMC Pediatr. 2021, 21, 18. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Harmatz, P.; Beck, M.; Jones, S.; Wood, T.; Lachman, R.; Gravance, C.G.; Orii, T.; Tomatsu, S. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol. Genet. Metab. 2013, 110, 54–64. [Google Scholar] [CrossRef]
- Morrone, A.; Caciotti, A.; Atwood, R.; Davidson, K.; Du, C.; Francis-Lyon, P.; Harmatz, P.; Mealiffe, M.; Mooney, S.; Oron, T.R.; et al. MorquioA Syndrome-Associated Mutations: A Review of Alterations in the GALNS Gene and a New Locus-Specific Database. Hum. Mutat. 2014, 35, 1271–1279. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Nishioka, T.; Gutierrez, M.A.; Peña, O.M.; Firescu, G.G.T.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512. [Google Scholar] [CrossRef]
- Yi, M.; Wang, Y.; Gao, X.; Han, L.; Qiu, W.; Gu, X.; Maegawa, G.H.B.; Zhang, H. Investigation of GALNS variants and genotype–phenotype correlations in a large cohort of patients with mucopolysaccharidosis type IVA. J. Inherit. Metab. Dis. 2022, 45, 593–604. [Google Scholar] [CrossRef]
- Caciotti, A.; Tonin, R.; Mort, M.; Cooper, D.N.; Gasperini, S.; Rigoldi, M.; Parini, R.; Deodato, F.; Taurisano, R.; Sibilio, M.; et al. Mis-splicing of the GALNS gene resulting from deep intronic mutations as a cause of Morquio a disease. BMC Med. Genet. 2018, 19, 183. [Google Scholar] [CrossRef]
- Jezela-Stanek, A.; Różdżyńska-Świątkowska, A.; Kulpanovich, A.; Ciara, E.; Marucha, J.; Tylki-Szymańska, A. Novel data on growth phenotype and causative genotypes in 29 patients with Morquio (Morquio-Brailsford) syndrome from Central-Eastern Europe. J. Appl. Genet. 2019, 60, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Frigeni, M.; Rodriguez-Buritica, D.F.; Saavedra, H.; Gunther, K.A.; Hillman, P.R.; Balaguru, D.; Northrup, H. The youngest pair of siblings with Mucopolysaccharidosis typeIVA to receive enzyme replacement therapy to date: A case report. Am. J. Med. Genet. Part A 2021, 185, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, E.; Deroma, L.; Hendriksz, C.J.; Hollak, C.; Krishan, A. Enzyme replacement therapy for mucopolysaccharidosis type IV (Morquio syndrome). Cochrane Database Syst. Rev. 2018, 2018, CD012961. [Google Scholar] [CrossRef]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef]
- Hendriksz, C.J. Elosulfase alfa (BMN 110) for the treatment of mucopolysaccharidosis IVA (Morquio A Syndrome). Expert Rev. Clin. Pharmacol. 2016, 9, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Alméciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodríguez-López, A.; Guzman, E.; Garzón, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. J. Med. Chem. 2019, 62, 6175–6189. [Google Scholar] [CrossRef]
- Sato, T.; Onai, N.; Yoshihara, H.; Arai, F.; Suda, T.; Ohteki, T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nat. Med. 2009, 15, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Akyol, M.U.; MPS Consensus Programme Steering Committee; Alden, T.D.; Amartino, H.; Ashworth, J.; Belani, K.; Berger, K.I.; Borgo, A.; Braunlin, E.; Eto, Y.; et al. Recommendations for the management of MPS IVA: Systematic evidence- and consensus-based guidance. Orphanet J. Rare Dis. 2019, 14, 137. [Google Scholar] [CrossRef]
- Sawamoto, K.; Gonzalez, J.V.A.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. [Google Scholar] [CrossRef]
- Mansoor, S. Trends of congenital hypothyroidism and inborn errors of metabolism in Pakistan. Orphanet J. Rare Dis. 2020, 15, 321. [Google Scholar] [CrossRef]
- Riaz, M.; Tiller, J.; Ajmal, M.; Azam, M.; Qamar, R.; Lacaze, P. Implementation of public health genomics in Pakistan. Eur. J. Hum. Genet. 2019, 27, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Management of Birth Defects and Haemoglobin Disorders. 2006; World Health Organization. Available online: https://apps.who.int/iris/bitstream/handle/10665/43587/9789241594929_eng.pdf?sequence=1&isAllowed=y (accessed on 10 December 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Severity | Mutation (p.Asp233Asn) Zygosity | Clinical Features of Patients | Mutated Allele (A) Count for c.697G>A (p.Asp233A)sn | Number of Individuals with Genotype | Allele Frequency Distribution | c.697G>A (p.Asp233Asn) Allele Frequency GnomAD | Study Type | Ethnicity | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Severe | Homozygous (p.Asp233Asn) | Spondylo-epiphyseal dysplasia, short stature, chest protrusion, short neck, vulgus deformity of the knees, scoliosis, and, abnormal gait | 7 | GG = 2 AA = 4 GA = 3 | 16.6% 33.3% 50% | 0.00001 | Familial | Pakistani | Current study |
| Severe | Homozygous (p.Asp233Asn) Compound Heterozygous (p.Asp233Asn p.Ala296Val) | Bone dysplasia, short trunk, and corneal clouding | 9 | GG = 422 AA = 8 GA = 1 | 97.7% 1.9% 0.46% | Case study | Chinese | [22] | |
| Severe | Compound heterozygous (p.Asp233Asn p.Lys153_Phe306del) | Spondyloepiphyseal dysplasia, chest deformity, coxa valga, hepatomegaly, hearing loss, and mental retardation | 1 | GG = 10 AA = 0 GA = 1 | 83.3% 0% 16.6% | Case study | Italian | [23] | |
| Mild | Compound heterozygous (p.Gly168Arg) (Asp233Asn) | Chest deformity | 1 | GG = 114 AA = 0 GA = 1 | 98.2% 0% 1.7% | Familial | Polish | [24] | |
| Severe | Homozygous (Asp233Asn) | Bone deformity, growth retardation, short stature, and cervical spine instability | 8 | GG = 488 AA = 8 GA = 0 | 98.3% 1.6% 0% | Familial | German, Polish, Indian | [17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghafoor, S.; Silveira, K.d.C.; Qamar, R.; Azam, M.; Kannu, P. Exome Sequencing Identifies a Biallelic GALNS Variant (p.Asp233Asn) Causing Mucopolysaccharidosis Type IVA in a Pakistani Consanguineous Family. Genes 2022, 13, 1743. https://doi.org/10.3390/genes13101743
Ghafoor S, Silveira KdC, Qamar R, Azam M, Kannu P. Exome Sequencing Identifies a Biallelic GALNS Variant (p.Asp233Asn) Causing Mucopolysaccharidosis Type IVA in a Pakistani Consanguineous Family. Genes. 2022; 13(10):1743. https://doi.org/10.3390/genes13101743
Chicago/Turabian StyleGhafoor, Saima, Karina da Costa Silveira, Raheel Qamar, Maleeha Azam, and Peter Kannu. 2022. "Exome Sequencing Identifies a Biallelic GALNS Variant (p.Asp233Asn) Causing Mucopolysaccharidosis Type IVA in a Pakistani Consanguineous Family" Genes 13, no. 10: 1743. https://doi.org/10.3390/genes13101743