

Robust Genetic Analysis of the X-Linked Anophthalmic (Ie) Mouse
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. High Molecular Weight DNA Isolation
2.3. Whole Genome Sequencing
2.4. Long-Read Sequencing
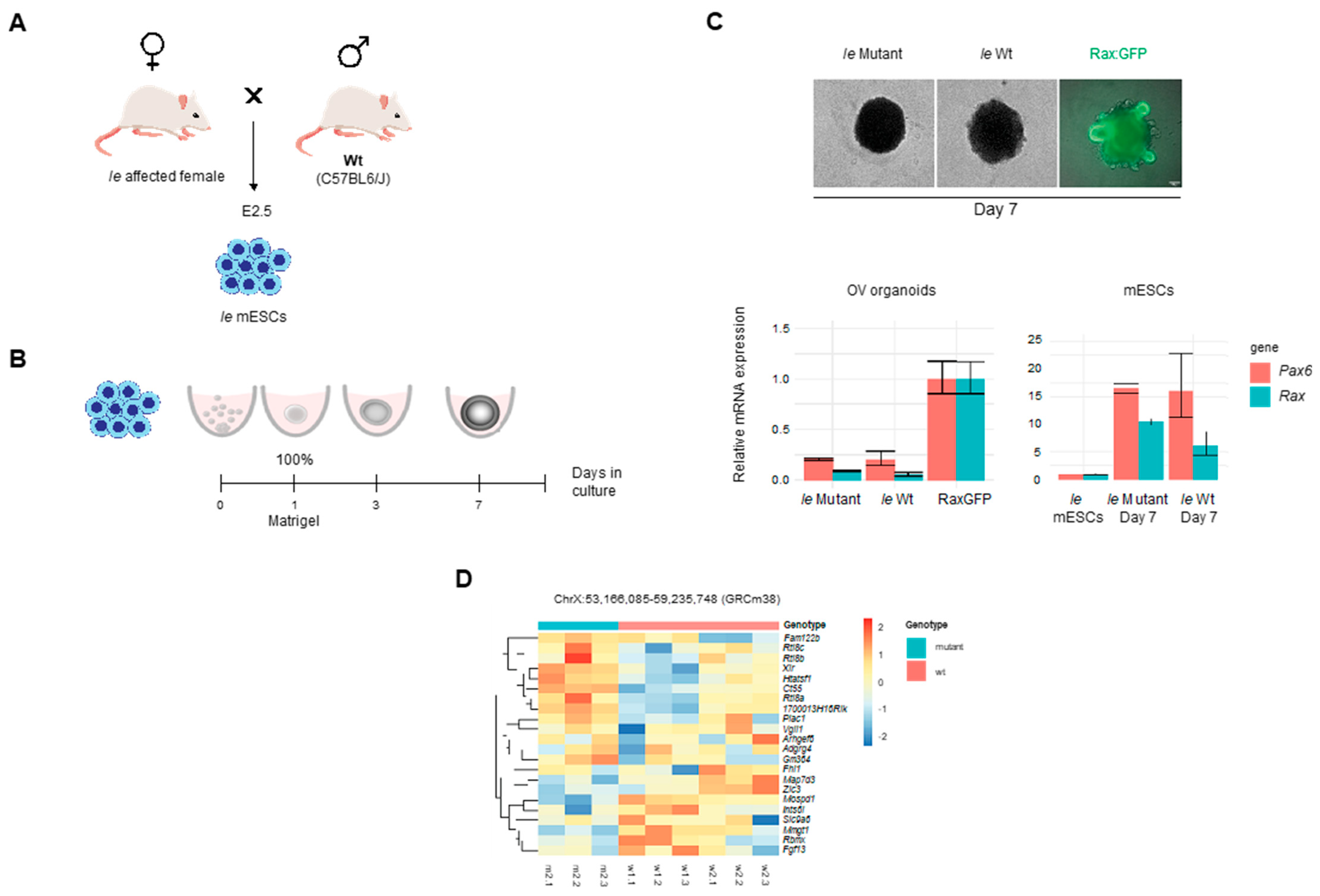
2.5. Derivation of mESCs
2.6. DNA Isolation from mESCs
2.7. Sex Determination and Genotyping of mESCs
2.8. Optic Organoids Culture
2.9. Statistical Analysis
2.10. RNA-seq
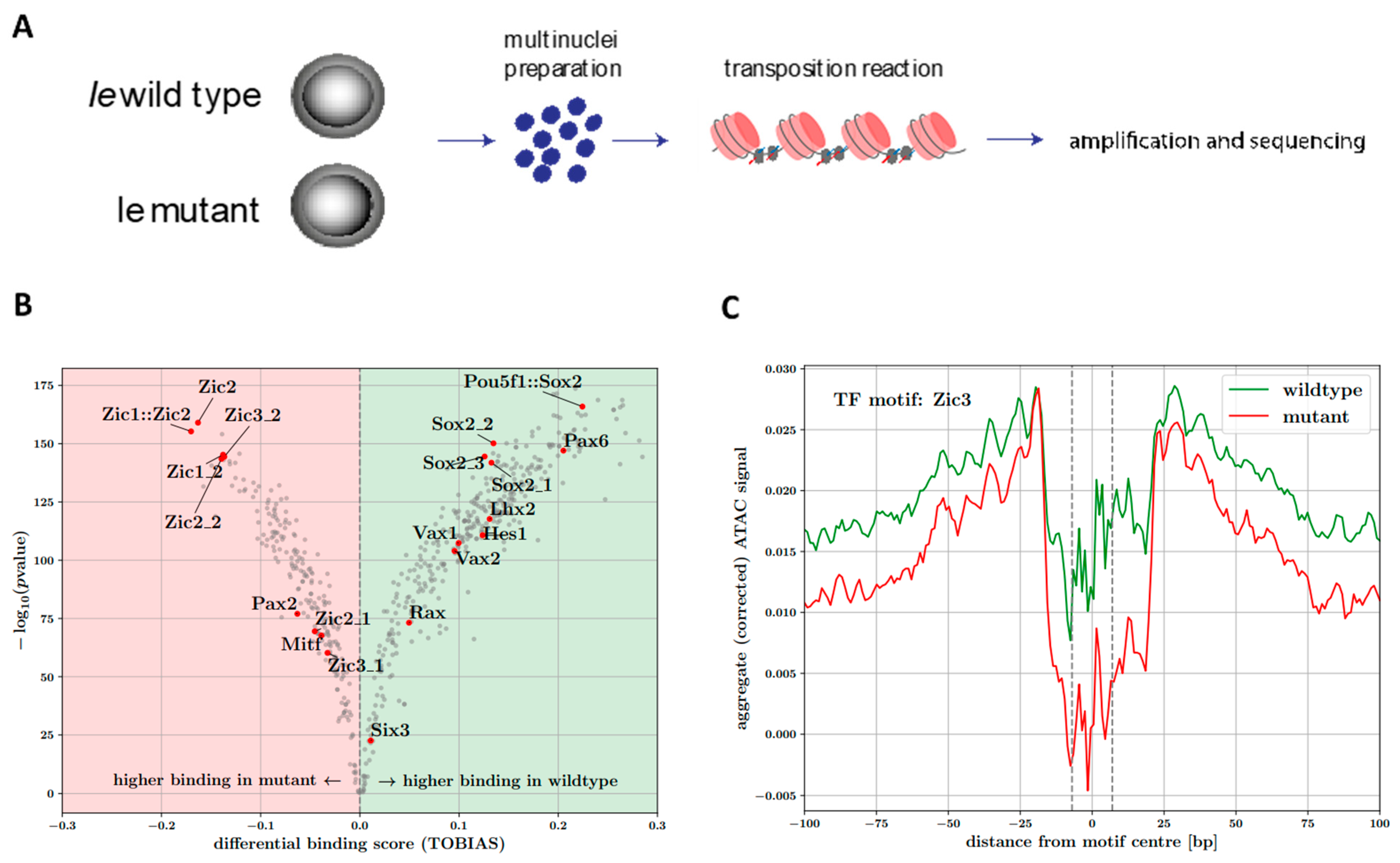
2.11. ATAC-seq
2.12. ATAC-seq Mapping and Analysis
3. Results
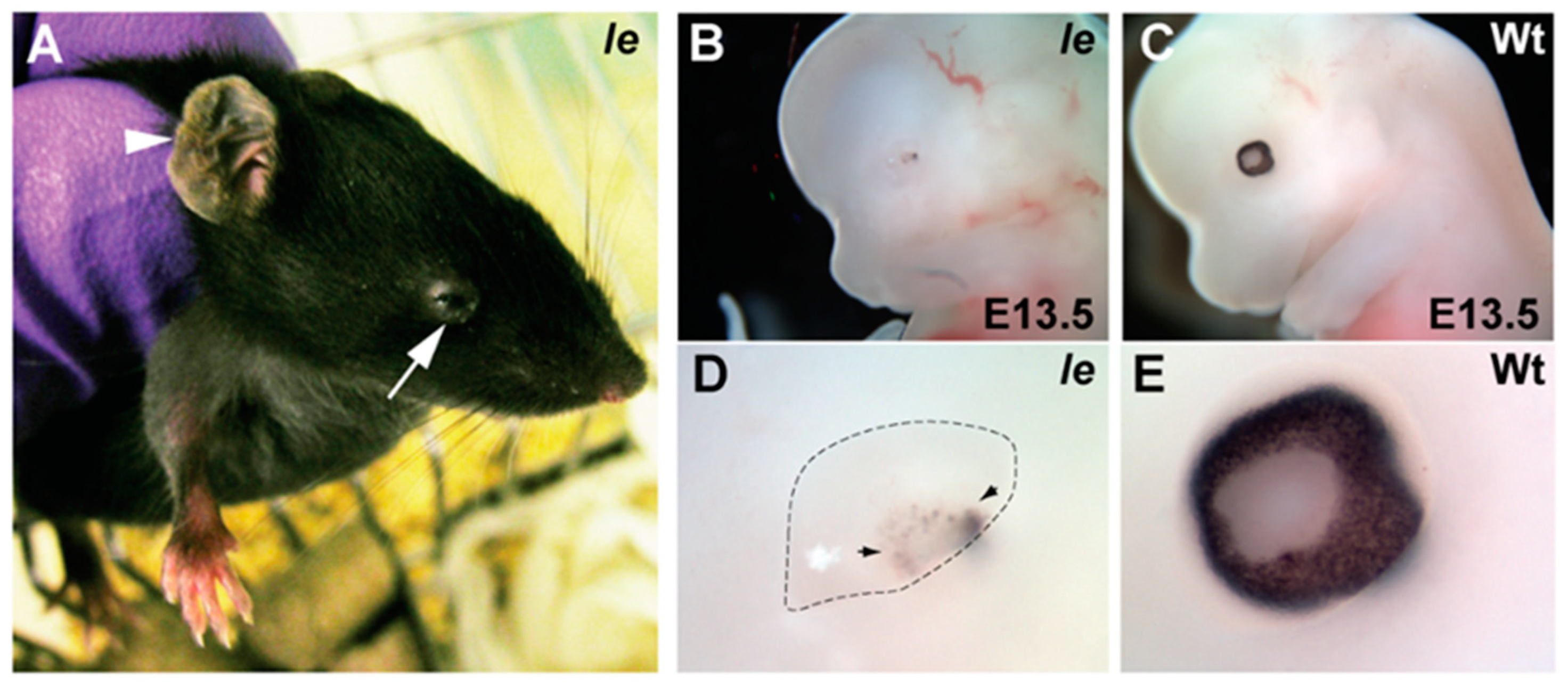
3.1. Phenotyping Ie Ocular Defects
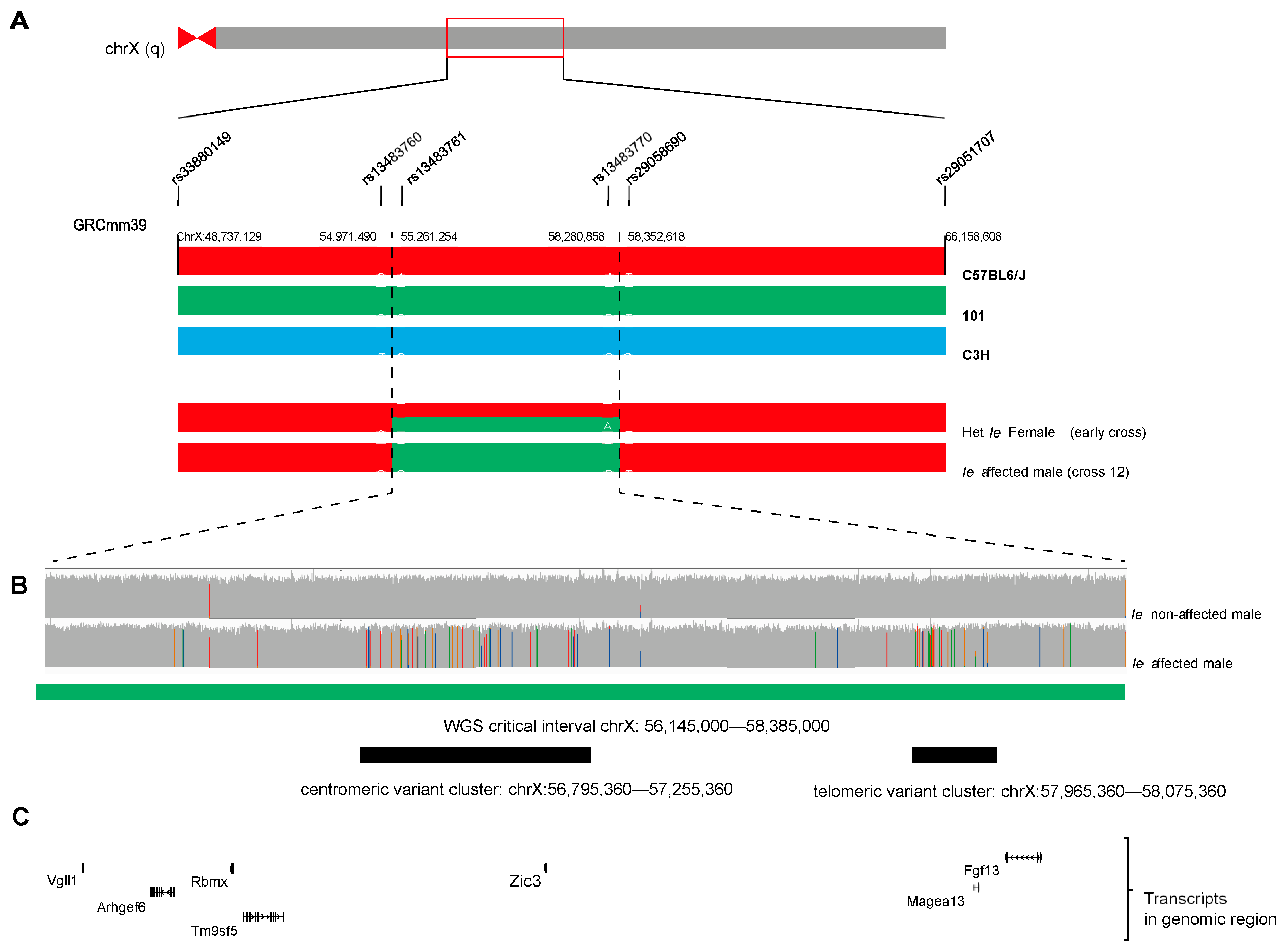
3.2. Genetic Mapping Ie
3.3. Whole Genome Sequencing
3.4. Long-Read Genomic DNA Sequencing
3.5. Expression Analyses
3.6. ATAC-seq
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Open Access
References
- Williamson, K.A.; FitzPatrick, D.R. The Genetic Architecture of Microphthalmia, Anophthalmia and Coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Kamachi, Y.; Matsunami, H.; Imada, K.; Uchikawa, M.; Kondoh, H. PAX6 and SOX2-Dependent Regulation of the Sox2 Enhancer N-3 Involved in Embryonic Visual System Development. Genes Cells 2007, 12, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Plaisancié, J.; Ceroni, F.; Holt, R.; Zazo Seco, C.; Calvas, P.; Chassaing, N.; Ragge, N.K. Genetics of Anophthalmia and Microphthalmia. Part 1: Non-Syndromic Anophthalmia/Microphthalmia. Hum. Genet. 2019, 138, 799–830. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.N.; Williamson, K.A.; Fitzpatrick, D.R. The Genetic Architecture of Aniridia and Gillespie Syndrome. Hum. Genet. 2018, 138, 881–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunsicker, P. Ie = Eye-Ear Reduction X Linked. Mouse News Lett. 1974, 50, 51–52. [Google Scholar]
- Garber, E.D.; D Garber, B.E. “Bent-Tail,” A Dominant, Sex-Linked Mutation in the Mouse. Proc. Natl. Acad. Sci. USA 1952, 38, 876–879. [Google Scholar] [CrossRef] [Green Version]
- Carrel, T.; Purandare, S.M.; Harrison, W.; Elder, F.; Fox, T.; Casey, B.; Herman, G.E. The X-Linked Mouse Mutation Bent Tail Is Associated with a Deletion of the Zic3 Locus. Hum. Mol. Genet. 2000, 9, 1937–1942. [Google Scholar] [CrossRef] [Green Version]
- Rainger, J.; Keighren, M.; Keene, D.R.; Charbonneau, N.L.; Rainger, J.K.; Fisher, M.; Mella, S.; Huang, J.T.J.; Rose, L.; van’t Hof, R.; et al. A Trans-Acting Protein Effect Causes Severe Eye Malformation in the Mp Mouse. PLOS Genet. 2013, 9, e1003998. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.; O’Connor, B. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse Genomic Variation and Its Effect on Phenotypes and Gene Regulation. Nature 2011, 477, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Coster, W.; De Rijk, P.; De Roeck, A.; De Pooter, T.; D’hert, S.; Strazisar, M.; Sleegers, K.; Van Broeckhoven, C. Structural Variants Identified by Oxford Nanopore PromethION Sequencing of the Human Genome. Genome Res. 2019, 29, 1178–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; Von Haeseler, A.; Schatz, M.C. Accurate Detection of Complex Structural Variations Using Single-Molecule Sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient Structural Variations Have Strong Effects on Quantitative Traits and Reproductive Isolation in Fission Yeast. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.; Jones, K. Derivation of Mouse Embryonic Stem (ES) Cell Lines Using Small-Molecule Inhibitors of Erk and Gsk3 Signaling (2i). Cold Spring Harb. Protoc. 2017, 2017, 379–386. [Google Scholar] [CrossRef]
- Tunster, S.J. Genetic Sex Determination of Mice by Simplex PCR. Biol. Sex Differ. 2017, 8, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Sasai, Y. Mouse Embryonic Stem Cell Culture for Generation of Three-Dimensional Retinal and Cortical Tissues. Nat. Protoc. 2012, 7, 69–79. [Google Scholar] [CrossRef]
- Ahmed, M.; Kim, D.R. Pcr: An R Package for Quality Assessment, Analysis and Testing of QPCR Data. PeerJ 2018, 6, e4473. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential Expression Analysis of Multifactor RNA-Seq Experiments with Respect to Biological Variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 2015, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The Nf-Core Framework for Community-Curated Bioinformatics Pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentsen, M.; Goymann, P.; Schultheis, H.; Klee, K.; Petrova, A.; Wiegandt, R.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; et al. ATAC-Seq Footprinting Unravels Kinetics of Transcription Factor Binding during Zygotic Genome Activation. Nat. Commun. 2020, 11, 4267. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; Van Der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. Emerging Roles of TEAD Transcription Factors and Its Coactivators in Cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Faucheux, C.; Naye, F.; Tréguer, K.; Fédou, S.; Thiébaud, P.; Thézé, N. Vestigial like Gene Family Expression in Xenopus: Common and Divergent Features with Other Vertebrates. Int. J. Dev. Biol. 2010, 54, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.; Gelbart, W.; Miller, J. Chromosomal Rearrangements. Mod. Genet. Anal. 1999, 112–118. [Google Scholar] [CrossRef]
- Patel, A.; Anderson, G.; Galea, G.L.; Balys, M.; Sowden, J.C. A Molecular and Cellular Analysis of Human Embryonic Optic Fissure Closure Related to the Eye Malformation Coloboma. Development 2020, 147, dev193649. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Furuta, A.; Setsuie, R.; Kikuchi, H.; Wang, Y.L.; Sakurai, M.; Kwon, J.; Noda, M.; Wada, K. Photoreceptor Cell Apoptosis in the Retinal Degeneration of Uchl3-Deficient Mice. Am. J. Pathol. 2006, 169, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-Throughput Discovery of Novel Developmental Phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.S.; FitzPatrick, D.R. Anophthalmia and Microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Adewoye, A.B.; Lindsay, S.J.; Dubrova, Y.E.; Hurles, M.E. The Genome-Wide Effects of Ionizing Radiation on Mutation Induction in the Mammalian Germline. Nat. Commun. 2015, 6, 6684. [Google Scholar] [CrossRef] [Green Version]
- Satoh, Y.; Asakawa, J.I.; Nishimura, M.; Kuo, T.; Shinkai, N.; Cullings, H.M.; Minakuchi, Y.; Sese, J.; Toyoda, A.; Shimada, Y.; et al. Characteristics of Induced Mutations in Offspring Derived from Irradiated Mouse Spermatogonia and Mature Oocytes. Sci. Rep. 2020, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Ware, S.M.; Harutyunyan, K.G.; Belmont, J.W. Zic3 Is Critical for Early Embryonic Patterning during Gastrulation. Dev. Dyn. 2006, 235, 776–785. [Google Scholar] [CrossRef]
- Hatayama, M.; Aruga, J. Role of Zic Family Proteins in Transcriptional Regulation and Chromatin Remodeling. Adv. Exp. Med. Biol. 2018, 1046, 353–380. [Google Scholar] [CrossRef]
- Yang, S.H.; Andrabi, M.; Biss, R.; Murtuza Baker, S.; Iqbal, M.; Sharrocks, A.D. ZIC3 Controls the Transition from Naive to Primed Pluripotency. Cell Rep. 2019, 27, 3215–3227.e6. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Moran, B.A.; Papanastasiou, A.S.; Parry, D.; Meynert, A.; Gautier, P.; Grimes, G.; Adams, I.R.; Trejo-Reveles, V.; Bengani, H.; Keighren, M.; et al. Robust Genetic Analysis of the X-Linked Anophthalmic (Ie) Mouse. Genes 2022, 13, 1797. https://doi.org/10.3390/genes13101797
Hernandez-Moran BA, Papanastasiou AS, Parry D, Meynert A, Gautier P, Grimes G, Adams IR, Trejo-Reveles V, Bengani H, Keighren M, et al. Robust Genetic Analysis of the X-Linked Anophthalmic (Ie) Mouse. Genes. 2022; 13(10):1797. https://doi.org/10.3390/genes13101797
Chicago/Turabian StyleHernandez-Moran, Brianda A., Andrew S. Papanastasiou, David Parry, Alison Meynert, Philippe Gautier, Graeme Grimes, Ian R. Adams, Violeta Trejo-Reveles, Hemant Bengani, Margaret Keighren, and et al. 2022. "Robust Genetic Analysis of the X-Linked Anophthalmic (Ie) Mouse" Genes 13, no. 10: 1797. https://doi.org/10.3390/genes13101797