
Influence of Parent-of-Origin on Intellectual Outcomes in the Chromosome 22q11.2 Deletion Syndrome

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Study Design
2.2. Analyses
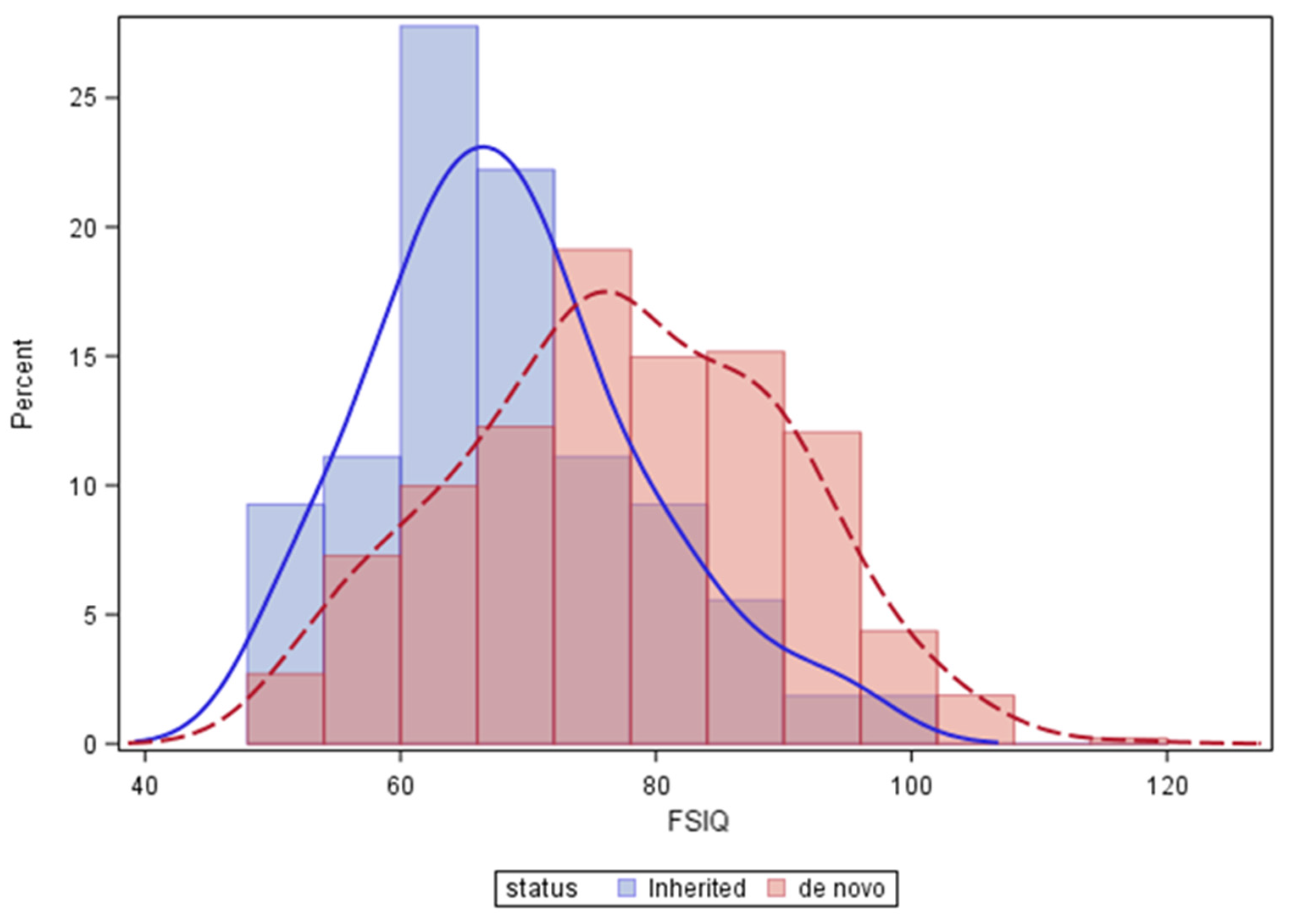
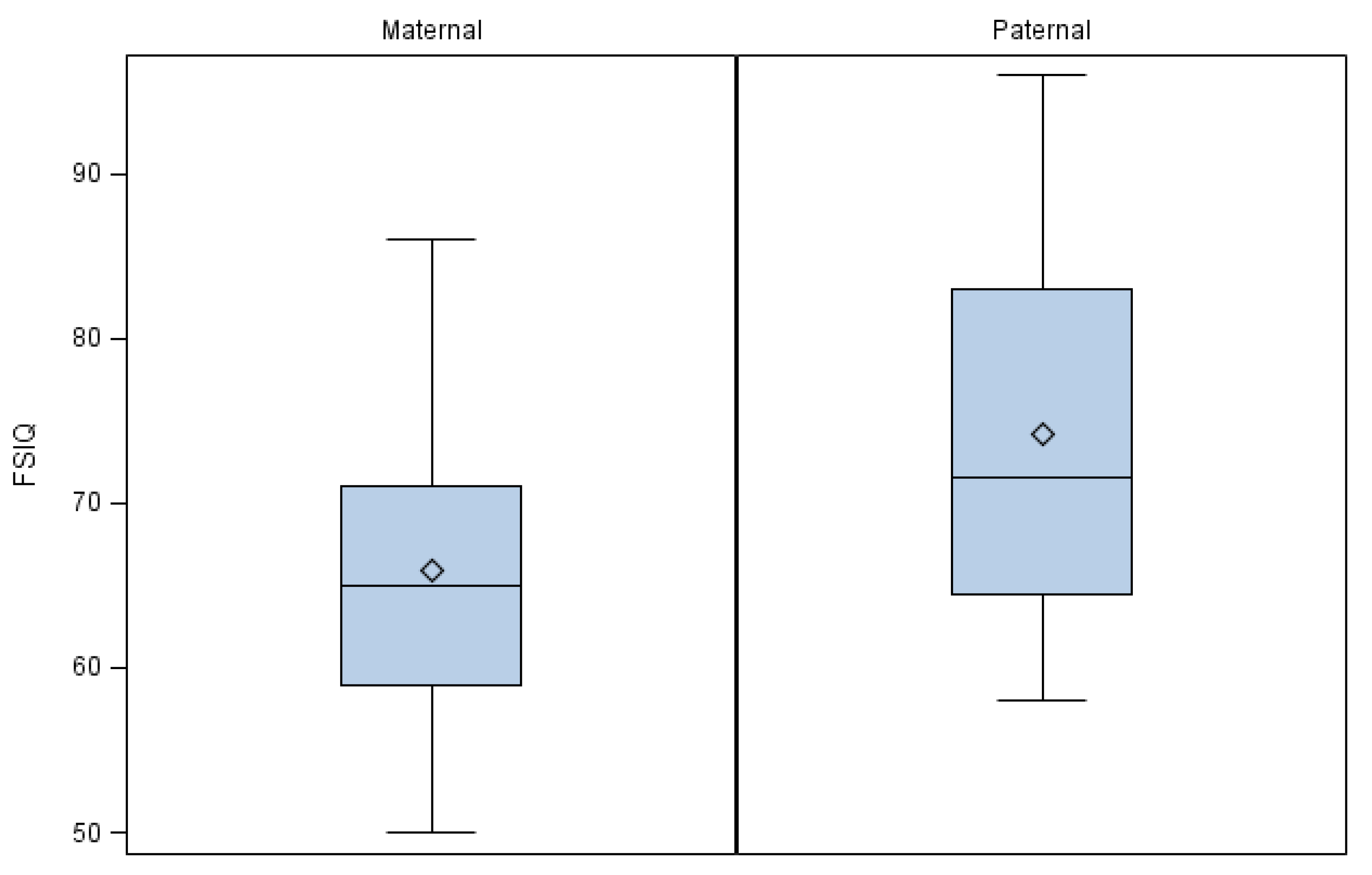
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the Contemporary Live-Birth Prevalence of Recurrent 22q11.2 Deletions: A Cross-Sectional Analysis from Population-Based Newborn Screening. CMAJ Open 2021, 9, E802–E809. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R.; Molina Gomes, D.; Ferreira, J.C.P.B.; Dupont, C.; Alesi, V.; Gouas, L.; Horelli-Kuitunen, N.; Choy, K.W.; García-Herrero, S.; de la Vega, A.G.; et al. Prevalence of Recurrent Pathogenic Microdeletions and Microduplications in over 9500 Pregnancies. Prenat Diagn 2015, 35, 801–809. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGinn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What Is New with 22q? An Update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet A 2018, 176, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Tonnesen, M.K.; Laufer-Cahana, A.; Finucane, B.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H. Phenotype of the 22q11.2 Deletion in Individuals Identified through an Affected Relative: Cast a Wide FISHing Net! Genet Med. 2001, 3, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scambler, P.J. The 22q11 Deletion Syndromes. Hum. Mol. Genet 2000, 9, 2421–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, J.C.; Crowley, T.B.; Jyonouchi, S.; Heimall, J.; Zackai, E.H.; Sullivan, K.E.; McDonald-McGinn, D.M. Identification of 22q11.2 Deletion Syndrome via Newborn Screening for Severe Combined Immunodeficiency. J. Clin. Immunol. 2017, 37, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; McDonald-McGinn, D.M.; Devriendt, K.; Digilio, M.C.; Goldenberg, P.; Habel, A.; Marino, B.; Oskarsdottir, S.; Philip, N.; Sullivan, K.; et al. Practical Guidelines for Managing Patients with 22q11.2 Deletion Syndrome. J. Pediatr. 2011, 159, 332–339.e1. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.D.; Butcher, N.J.; Boot, E.; Hodgkinson, K.A.; Heung, T.; Chow, E.W.C.; Guna, A.; Crowley, T.B.; Zackai, E.; McDonald-McGinn, D.M.; et al. Elucidating the Diagnostic Odyssey of 22q11.2 Deletion Syndrome. Am. J. Med. Genet A 2018, 176, 936–944. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, B.; Devriendt, K.; Fryns, J.-P.; Vogels, A.; Gewillig, M.; Swillen, A. Intellectual Abilities in a Large Sample of Children with Velo-Cardio-Facial Syndrome: An Update. J. Intellect. Disabil. Res. 2007, 51, 666–670. [Google Scholar] [CrossRef]
- Swillen, A.; Devriendt, K.; Legius, E.; Eyskens, B.; Dumoulin, M.; Gewillig, M.; Fryns, J.P. Intelligence and Psychosocial Adjustment in Velocardiofacial Syndrome: A Study of 37 Children and Adolescents with VCFS. J. Med. Genet 1997, 34, 453–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swillen, A.; McDonald-McGinn, D. Developmental Trajectories in 22q11.2 Deletion. Am. J. Med. Genet C Semin Med. Genet 2015, 169, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Guo, T.; Fiksinski, A.; Breetvelt, E.; McDonald-McGinn, D.M.; Crowley, T.B.; Diacou, A.; Schneider, M.; Eliez, S.; Swillen, A.; et al. Variance of IQ Is Partially Dependent on Deletion Type among 1427 22q11.2 Deletion Syndrome Subjects. Am. J. Med. Genet A 2018, 176, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, D.; Presburger, G.; Levy, D.; Nahmani, A.; Burg, M.; Berant, M.; Blieden, L.C.; Finkelstein, Y.; Frisch, A.; Apter, A.; et al. Genetic, Developmental, and Physical Factors Associated with Attention Deficit Hyperactivity Disorder in Patients with Velocardiofacial Syndrome. Am. J. Med. Genet B Neuropsychiatr. Genet. 2004, 126B, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Grand, K.; Levitt Katz, L.E.; Crowley, T.B.; Moss, E.; Lessig, M.; Bamba, V.; Lord, K.; Zackai, E.H.; Emanuel, B.S.; Valverde, K.; et al. The Impact of Hypocalcemia on Full Scale IQ in Patients with 22q11.2 Deletion Syndrome. Am. J. Med. Genet A 2018, 176, 2167–2171. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.J.; Tang, S.X.; McDonald-McGinn, D.M.; Calkins, M.E.; Whinna, D.A.; Souders, M.C.; Zackai, E.H.; Goldmuntz, E.; Gaynor, J.W.; Gur, R.C.; et al. Contribution of Congenital Heart Disease to Neuropsychiatric Outcome in School-Age Children with 22q11.2 Deletion Syndrome. Am. J. Med. Genet B Neuropsychiatr. Genet. 2014, 165B, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, A.S.; Marshall, C.R.; Lionel, A.C.; Chow, E.W.C.; Scherer, S.W. Copy Number Variations and Risk for Schizophrenia in 22q11.2 Deletion Syndrome. Hum. Mol. Genet 2008, 17, 4045–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiksinski, A.M.; Heung, T.; Corral, M.; Breetvelt, E.J.; Costain, G.; Marshall, C.R.; Kahn, R.S.; Vorstman, J.A.S.; Bassett, A.S. Within-Family Influences on Dimensional Neurobehavioral Traits in a High-Risk Genetic Model. Psychol. Med. 2021, 1–9, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Swillen, A.; Moss, E.; Duijff, S. Neurodevelopmental Outcome in 22q11.2 Deletion Syndrome and Management. Am. J. Med. Genet A 2018, 176, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Breetvelt, E.J.; Duijff, S.N.; Eliez, S.; Schneider, M.; Jalbrzikowski, M.; Armando, M.; Vicari, S.; Shashi, V.; Hooper, S.R.; et al. Cognitive Decline Preceding the Onset of Psychosis in Patients with 22q11.2 Deletion Syndrome. JAMA Psychiatry 2015, 72, 377–385. [Google Scholar] [CrossRef]
- Gothelf, D.; Aviram-Goldring, A.; Burg, M.; Steinberg, T.; Mahajnah, M.; Frisch, A.; Fennig, S.; Zalsman, G.; Weizman, A. Cognition, Psychosocial Adjustment and Coping in Familial Cases of Velocardiofacial Syndrome. J. Neural Transm 2007, 114, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Delio, M.; Guo, T.; McDonald-McGinn, D.M.; Zackai, E.; Herman, S.; Kaminetzky, M.; Higgins, A.M.; Coleman, K.; Chow, C.; Jarlbrzkowski, M.; et al. Enhanced Maternal Origin of the 22q11.2 Deletion in Velocardiofacial and DiGeorge Syndromes. Am. J. Hum. Genet. 2013, 92, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Costain, G.; Chow, E.W.C.; Silversides, C.K.; Bassett, A.S. Sex Differences in Reproductive Fitness Contribute to Preferential Maternal Transmission of 22q11.2 Deletions. J. Med. Genet 2011, 48, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, E.M.; Batshaw, M.L.; Solot, C.B.; Gerdes, M.; McDonald-McGinn, D.M.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H.; Wang, P.P. Psychoeducational Profile of the 22q11.2 Microdeletion: A Complex Pattern. J. Pediatr. 1999, 134, 193–198. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Variables | Linear Regression AnalysisInherited vs. de novo 22q11.2 Deletion (n = 535) a | Linear Regression AnalysisPaternally vs. Maternally Inherited 22q11.2 Deletion (n = 54) b | ||||||
|---|---|---|---|---|---|---|---|---|
| Β | 95% CI | p | Β | 95% CI | p | |||
| Inherited (vs. de novo) 22q11.2 deletion a | −8.11 | −11.61 | −4.62 | <0.0001 | ||||
| Paternal (vs. maternal) affected parent b | 8.17 | 2.44 | 13.89 | 0.0061 | ||||
| Age at IQ testing | −0.02 | −0.03 | −0.003 | 0.0177 | 0.05 | 0.003 | 0.10 | 0.0370 |
| Cohort site | −1.06 | −2.20 | 0.07 | 0.0655 | −5.05 | −8.96 | −1.14 | 0.0124 |
| Male sex | −0.95 | −3.04 | 1.15 | 0.3776 | −0.33 | 0.90 | −5.84 | 0.9021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGinn, D.E.; Crowley, T.B.; Heung, T.; Tran, O.; Moss, E.; Zackai, E.H.; Emanuel, B.S.; Chow, E.W.C.; Morrow, B.E.; Swillen, A.; et al. Influence of Parent-of-Origin on Intellectual Outcomes in the Chromosome 22q11.2 Deletion Syndrome. Genes 2022, 13, 1800. https://doi.org/10.3390/genes13101800
McGinn DE, Crowley TB, Heung T, Tran O, Moss E, Zackai EH, Emanuel BS, Chow EWC, Morrow BE, Swillen A, et al. Influence of Parent-of-Origin on Intellectual Outcomes in the Chromosome 22q11.2 Deletion Syndrome. Genes. 2022; 13(10):1800. https://doi.org/10.3390/genes13101800
Chicago/Turabian StyleMcGinn, Daniel E., T. Blaine Crowley, Tracy Heung, Oanh Tran, Edward Moss, Elaine H. Zackai, Beverly S. Emanuel, Eva W. C. Chow, Bernice E. Morrow, Ann Swillen, and et al. 2022. "Influence of Parent-of-Origin on Intellectual Outcomes in the Chromosome 22q11.2 Deletion Syndrome" Genes 13, no. 10: 1800. https://doi.org/10.3390/genes13101800
APA StyleMcGinn, D. E., Crowley, T. B., Heung, T., Tran, O., Moss, E., Zackai, E. H., Emanuel, B. S., Chow, E. W. C., Morrow, B. E., Swillen, A., Bassett, A. S., & McDonald-McGinn, D. M. (2022). Influence of Parent-of-Origin on Intellectual Outcomes in the Chromosome 22q11.2 Deletion Syndrome. Genes, 13(10), 1800. https://doi.org/10.3390/genes13101800