


Systematic Identification and Validation of Suitable Reference Genes for the Normalization of Gene Expression in Prunella vulgaris under Different Organs and Spike Development Stages



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Total RNA Extraction and cDNA Synthesis
2.3. Selection and Validation of Candidate RGs and the Design of qPCR Primers
2.4. Quantitative Real-Time PCR (qRT-PCR) Analyses
2.5. Analysis of Expression Stability of Candidate RGs
2.6. RGs Validation
2.7. Data Analysis
3. Results
3.1. Selection of RGs and Analysis of Primer Amplification Specificity and Efficiency
3.2. Expression Profiles of Candidate RGs
3.3. Expression Stability Estimation of Candidate RGs by Five Bioinformatic Programs
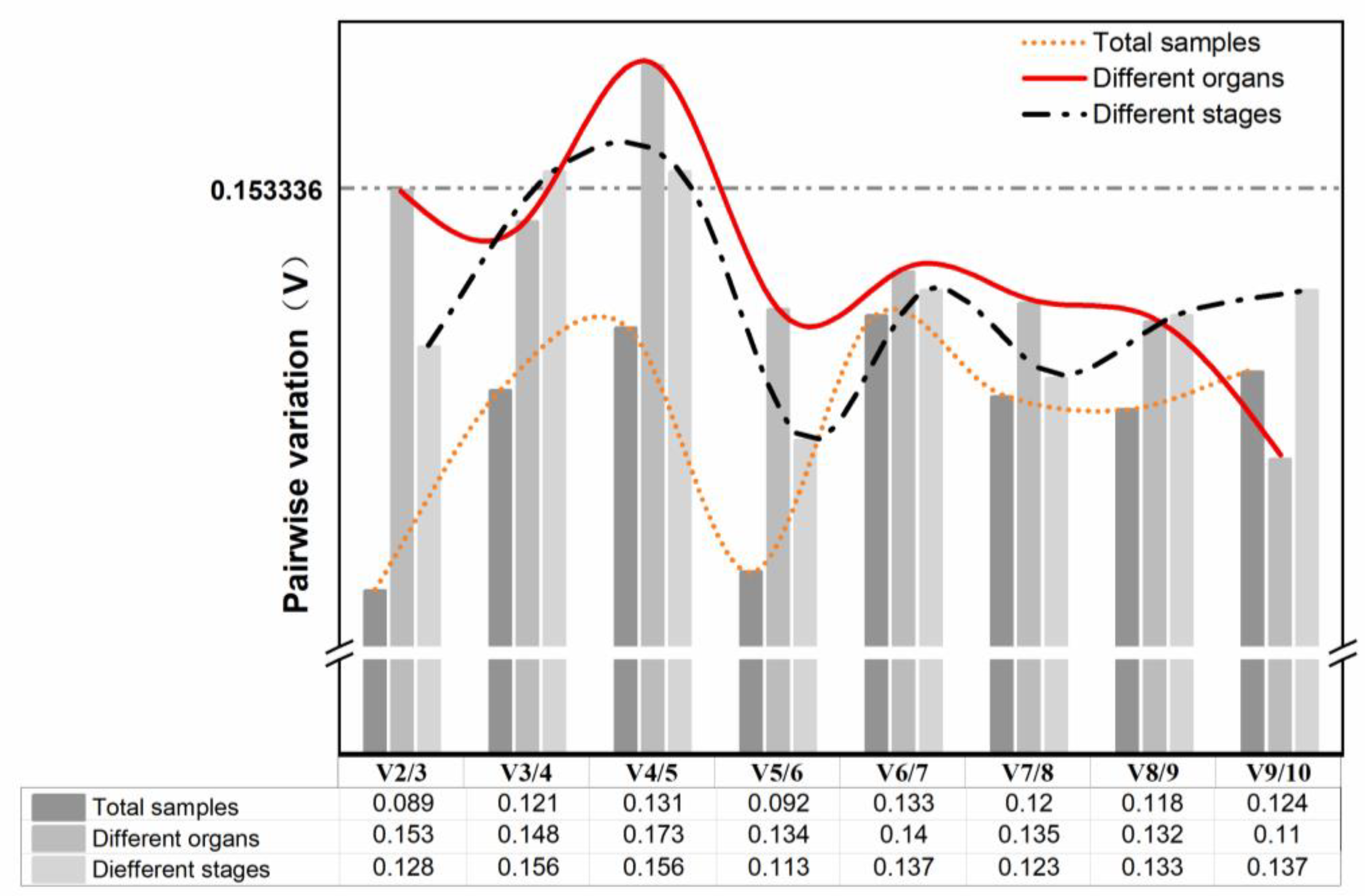
3.3.1. geNorm Analysis
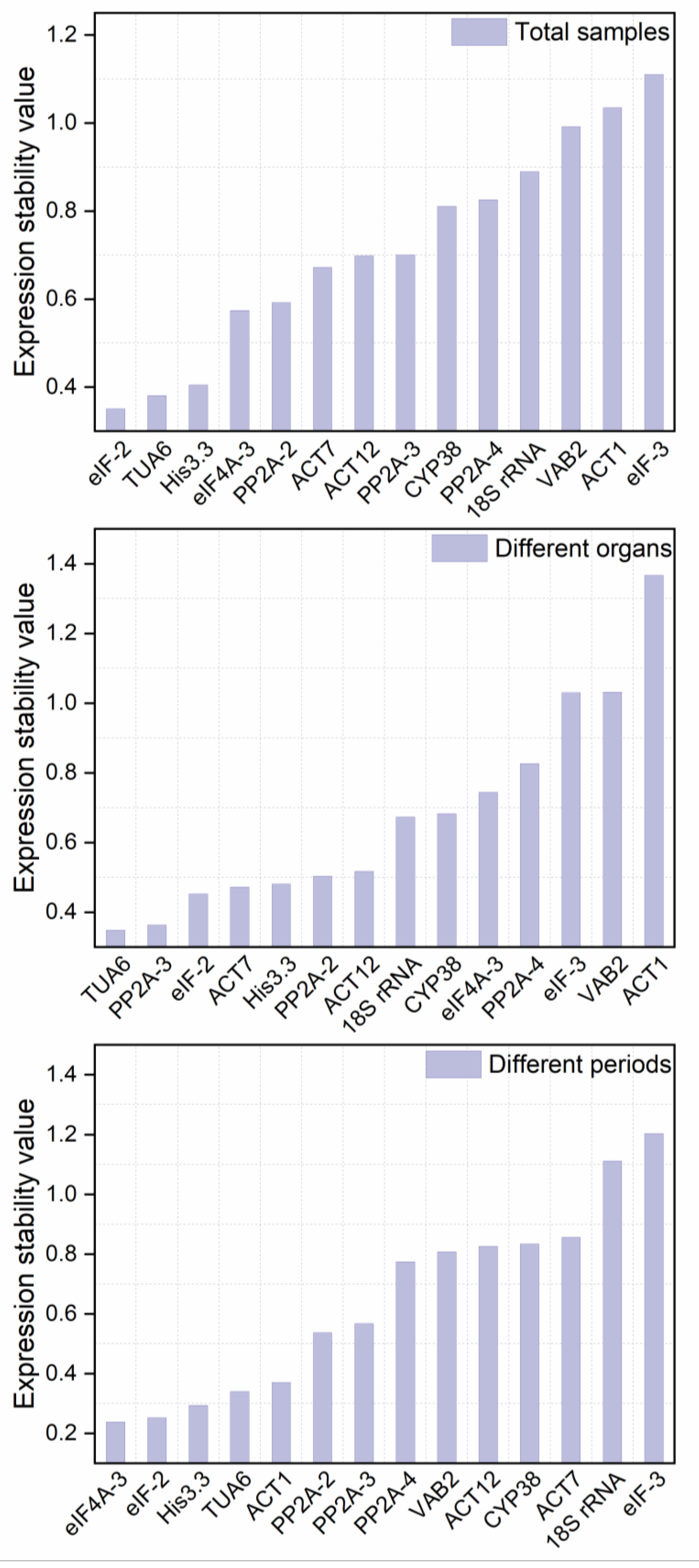
3.3.2. NormFinder Analysis
3.3.3. BestKeeper Analysis
3.3.4. Delta Ct Analysis
3.3.5. RefFinder Analysis
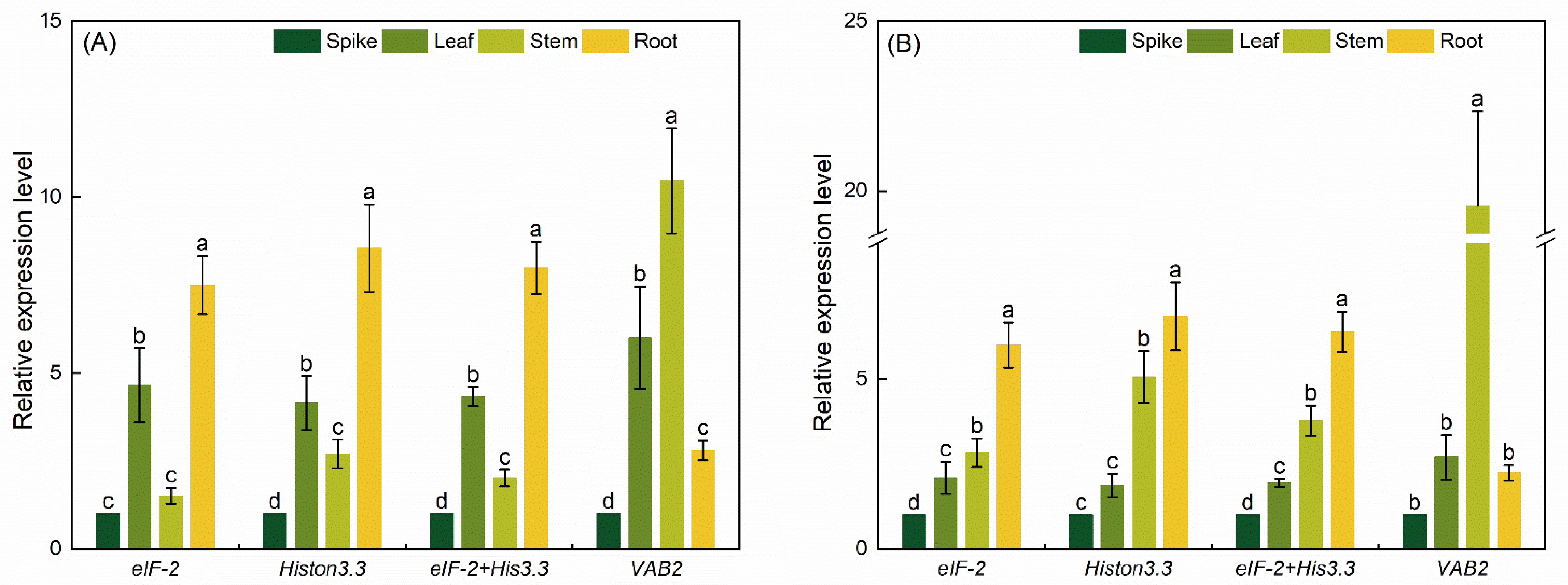
3.4. RGs’ Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Z.; Zhang, R.; Zhou, Z. Identification and Validation of Reference Genes for Gene Expression Analysis in Schima superba. Genes 2021, 12, 732. [Google Scholar] [CrossRef]
- Xu, L.; Xu, H.; Cao, Y.; Yang, P.; Feng, Y.; Tang, Y.; Yuan, S.; Ming, J. Validation of Reference Genes for Quantitative Real-Time PCR during Bicolor Tepal Development in Asiatic Hybrid Lilies (Lilium spp.). Front. Plant Sci. 2017, 8, 669. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.S.; Ahmed, R.; Haque, M.S.; Alam, M.M.; Islam, M.S. Identification and validation of reference genes for real-time quantitative RT-PCR analysis in jute. BMC Mol. Biol. 2019, 20, 13. [Google Scholar] [CrossRef]
- Deng, Y.; Li, Y.; Sun, H. Selection of reference genes for RT-qPCR normalization in blueberry (Vaccinium corymbosum × angustifolium) under various abiotic stresses. FEBS Open Bio 2020, 10, 1418–1435. [Google Scholar] [CrossRef]
- Kumar, D.; Das, P.K.; Sarmah, B.K. Reference gene validation for normalization of RT-qPCR assay associated with germination and survival of rice under hypoxic condition. J. Appl. Genet. 2018, 59, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Gao, Y.; Wáng, Y.; Wu, Y.; Wāng, Y.; Zhao, Y.; Chen, H.; Bao, D.; Xu, J.; Bian, X. Selection and Evaluation of Appropriate Reference Genes for RT-qPCR Normalization of Volvariella volvacea Gene Expression under Different Conditions. BioMed Res. Int. 2018, 2018, 6125706. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xie, W.; Yu, X.; Zhang, Z.; Zhao, Y.; Wang, N.; Wang, Y. Selection of Suitable Reference Genes for RT-qPCR Gene Expression Analysis in Siberian Wild Rye (Elymus sibiricus) under Different Experimental Conditions. Genes 2019, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Duan, H.; Chong, P.; Su, S.; Shan, L.; Yi, D.; Wang, L.; Li, Y. Systematic selection and validation of suitable reference genes for quantitative real-time PCR normalization studies of gene expression in Nitraria tangutorum. Sci. Rep. 2020, 10, 15891. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, A.; Yang, S.; Huang, L. Reference gene selection for real-time quantitative PCR normalization in Hemarthria compressa and Hemarthria altissima leaf tissue. Mol. Biol. Rep. 2019, 46, 4763–4769. [Google Scholar] [CrossRef]
- Chen, C.; Yuan, M.; Song, J.; Liu, Y.; Xia, Z.; Yuan, Y.; Wang, W.; Xie, Q.; Guan, X.; Chen, Q.; et al. Genome-wide identification and testing of superior reference genes for transcript normalization during analyses of flesh development in Asian pear cultivars. Sci. Hortic. 2020, 271, 109459. [Google Scholar] [CrossRef]
- Vera Hernández, F.P.; Martínez Núñez, M.; Ruiz Rivas, M.; Vázquez Portillo, R.E.; Bibbins Martínez, M.D.; Luna Suárez, S.; Rosas Cárdenas, F.F. Reference genes for RT-qPCR normalisation in different tissues, developmental stages and stress conditions of amaranth. Plant Biol. 2018, 20, 713–721. [Google Scholar] [CrossRef]
- Lucho, S.R.; do Amaral, M.N.; Benitez, L.C.; Milech, C.; Kleinowski, A.M.; Bianchi, V.J.; Braga, E.J.B. Validation of reference genes for RT-qPCR studies in Stevia rebaudiana in response to elicitor agents. Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2018, 24, 767–779. [Google Scholar] [CrossRef]
- Dixit, S.; Jangid, V.K.; Grover, A. Evaluation of suitable reference genes in Brassica juncea and its wild relative Camelina sativa for qRT-PCR analysis under various stress conditions. PLoS ONE 2019, 14, e0222530. [Google Scholar] [CrossRef]
- Jia, D.-H.; Wang, B.; Li, X.-L.; Tan, W.; Gan, B.-C.; Peng, W.-H. Validation of reference genes for quantitative gene expression analysis in Auricularia cornea. J. Microbiol. Methods 2019, 163, 105658. [Google Scholar] [CrossRef]
- Mo, J.; Xu, J.; Jin, W.; Yang, L.; Yin, T.; Shi, J. Identification of Reference Genes for Quantitative Gene Expression Studies in Pinus massoniana and Its Introgression Hybrid. Forests 2019, 10, 787. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhong, Y.; Bao, Z.; Wang, W.; Xu, X.; Gai, Y.; Wu, J. Evaluation of Angelica decursiva reference genes under various stimuli for RT-qPCR data normalization. Sci. Rep. 2021, 11, 18993. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, F.; Shi, D.; Wang, Y.; Xu, F.; Zeng, S. Selection and validation of reference genes for RT-qPCR analysis in Desmodium styracifolium Merr. 3 Biotech 2021, 11, 403. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Yang, H.; Lyu, L.; Li, W.; Wu, W. Selection and Validation of Candidate Reference Genes for Gene Expression Analysis by RT-qPCR in Rubus. Int. J. Mol. Sci. 2021, 22, 10533. [Google Scholar] [CrossRef]
- Qu, R.; Miao, Y.; Cui, Y.; Cao, Y.; Zhou, Y.; Tang, X.; Yang, J.; Wang, F. Selection of reference genes for the quantitative real-time PCR normalization of gene expression in Isatis indigotica fortune. BMC Mol. Biol. 2019, 20, 9. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, Research0034. [Google Scholar] [CrossRef] [Green Version]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Doddaraju, P.; Kumar, P.; Dashyal, M.S.; Girigowda, M. Identification of suitable reference genes for expression studies in pomegranate under different biotic and abiotic stress conditions. Mol. Biol. Rep. 2021, 48, 3935–3943. [Google Scholar] [CrossRef]
- Joseph, J.T.; Poolakkalody, N.J.; Shah, J.M. Plant reference genes for development and stress response studies. J. Biosci. 2018, 43, 173–187. [Google Scholar] [CrossRef]
- Fan, L.; Chen, M.; Dong, B.; Wang, N.; Yu, Q.; Wang, X.; Xuan, L.; Wang, Y.; Zhang, S.; Shen, Y. Transcriptomic Analysis of Flower Bud Differentiation in Magnolia sinostellata. Genes 2018, 9, 212. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Xia, B.; Xie, W.; Zhou, Y.; Xie, J.; Li, H.; Liao, D.; Lin, L.; Li, C. Phytochemistry and pharmacological activities of the genus Prunella. Food Chem. 2016, 204, 483–496. [Google Scholar] [CrossRef]
- Song, Y.-G.; Kang, L.; Tian, S.; Cui, L.-L.; Li, Y.; Bai, M.; Fang, X.-Y.; Cao, L.-H.; Coleman, K.; Miao, M.-S. Study on the anti-hepatocarcinoma effect and molecular mechanism of Prunella vulgaris total flavonoids. J. Ethnopharmacol. 2021, 273, 113891. [Google Scholar] [CrossRef]
- Fang, X.; Yu, M.M.; Yuen, W.H.; Zee, S.Y.; Chang, R.C. Immune modulatory effects of Prunella vulgaris L. on monocytes/macrophages. Int. J. Mol. Med. 2005, 16, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Brindley, M.A.; Widrlechner, M.P.; McCoy, J.A.; Murphy, P.; Hauck, C.; Rizshsky, L.; Nikolau, B.; Maury, W. Inhibition of lentivirus replication by aqueous extracts of Prunella vulgaris. Virol. J. 2009, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Huang, Q.; Fu, X.; Yue, X.J.; Liu, R.H.; You, L.J. Characterization, antioxidant and immunomodulatory activities of polysaccharides from Prunella vulgaris Linn. Int. J. Biol. Macromol. 2015, 75, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Lee, E.J.; Kim, H.R.; Hwang, K.A. In vitro antioxidant and anticancer effects of solvent fractions from Prunella vulgaris var. lilacina. BMC Complement. Altern. Med. 2013, 13, 310. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Huang, Q.; Xiao, J.; Fu, X.; You, L.; Liu, R.H. Preparation of Prunella vulgaris polysaccharide-zinc complex and its antiproliferative activity in HepG2 cells. Int. J. Biol. Macromol. 2016, 91, 671–679. [Google Scholar] [CrossRef]
- Raafat, K.; Wurglics, M.; Schubert-Zsilavecz, M. Prunella vulgaris L. active components and their hypoglycemic and antinociceptive effects in alloxan-induced diabetic mice. Biomed. Pharmacother. 2016, 84, 1008–1018. [Google Scholar] [CrossRef]
- Wang, S.J.; Wang, X.H.; Dai, Y.Y.; Ma, M.H.; Rahman, K.; Nian, H.; Zhang, H. Prunella vulgaris: A Comprehensive Review of Chemical Constituents, Pharmacological Effects and Clinical Applications. Curr. Pharm. Des. 2019, 25, 359–369. [Google Scholar] [CrossRef]
- Guo, Q. Study on Biological Characters and Active Components of SP6 Generation of Prunella vulgaris L. Master’s Thesis, Northwest A&F University, Xianyang, China, 2019. [Google Scholar]
- Radonić, A.; Thulke, S.; Mackay, I.M.; Landt, O.; Siegert, W.; Nitsche, A. Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2004, 313, 856–862. [Google Scholar] [CrossRef]
- Li, J.; Han, X.; Wang, C.; Qi, W.; Zhang, W.; Tang, L.; Zhao, X. Validation of Suitable Reference Genes for RT-qPCR Data in Achyranthes bidentata Blume under Different Experimental Conditions. Front. Plant Sci. 2017, 8, 776. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Gao, Z.; Li, H.; Li, Q.; Zhang, C.; Xu, W.; Song, S.; Ma, C.; Wang, S. Selection of reference genes for miRNA qRT-PCR under abiotic stress in grapevine. Sci. Rep. 2018, 8, 4444. [Google Scholar] [CrossRef]
- Cheng, T.; Zhu, F.; Sheng, J.; Zhao, L.; Zhou, F.; Hu, Z.; Diao, Y.; Jin, S. Selection of suitable reference genes for quantitive real-time PCR normalization in Miscanthus lutarioriparia. Mol. Biol. Rep. 2019, 46, 4545–4553. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Ma, J.; Wang, J.; Wu, X.; Li, P.; Yao, Y. Validation of suitable reference genes for gene expression analysis in the halophyte Salicornia europaea by real-time quantitative PCR. Front. Plant Sci. 2014, 5, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, M.; Wang, K.; Bai, Z.; Peng, L.; He, S.; Wang, Y.; Liang, Z. A tyrosine aminotransferase involved in rosmarinic acid biosynthesis in Prunella vulgaris L. Sci. Rep. 2017, 7, 4892. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Shin, Y.; Tuan, P.A.; Li, X.; Park, Y.; Park, N.I.; Park, S.U. Molecular cloning and characterization of genes involved in rosmarinic acid biosynthesis from Prunella vulgaris. Biol. Pharm. Bull. 2014, 37, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, X.; Shi, Q.; Yang, T.; Fei, Z.; Wei, A. Expression Stabilities of Ten Candidate Reference Genes for RT-qPCR in Zanthoxylum bungeanum Maxim. Molecules 2018, 23, 802. [Google Scholar] [CrossRef] [Green Version]
- Archer, M.; Xu, J. Current Practices for Reference Gene Selection in RT-qPCR of Aspergillus: Outlook and Recommendations for the Future. Genes 2021, 12, 960. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, H.; Chen, M.; Song, X.; Yu, C.; Zhao, Y.; Wu, Y. Reference Gene Selection for Quantitative Real-Time PCR of Mycelia from Lentinula edodes under High-Temperature Stress. BioMed Res. Int. 2018, 2018, 1670328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shen, T.; Zhou, X.; Tang, X.; Gao, R.; Xu, L.; Wang, L.; Zhou, Z.; Lin, J.; Hu, Y. Network pharmacology based virtual screening of active constituents of Prunella vulgaris L. and the molecular mechanism against breast cancer. Sci. Rep. 2020, 10, 15730. [Google Scholar] [CrossRef]
- Guo, Q.; Qu, H.; Zhang, H.; Zhong, X. Prunella vulgaris L. Attenuates Experimental Autoimmune Thyroiditis by Inhibiting HMGB1/TLR9 Signaling. Drug Des. Dev. Ther. 2021, 15, 4559–4574. [Google Scholar] [CrossRef]
- Die, J.V.; Román, B.; Nadal, S.; González-Verdejo, C.I. Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta 2010, 232, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Su, Y.; Sun, J.; Liu, J.; Li, Z.; Zhang, B. Selection of stable reference genes for gene expression analysis in sweet potato (Ipomoea batatas L.). Mol. Cell. Probes 2020, 53, 101610. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Abbreviation | Gene ID | Gene Name | Arabidopsis Homolog Locus | Primer Sequences (5′–3′) (Forward/Reverse) | Tm (°C) | Length (bp) | Efficiency | R2 |
|---|---|---|---|---|---|---|---|---|
| ACT7 | SRR7873856.1.862141 | Actin | AT5G09810 | GTTACGAGCTTCCCGATGGA | 59.54 | 193 | 105.56% | 0.9989 |
| GATCCACCACTGAGCACGAT | 59.82 | |||||||
| ACT12 | SRR7873856.1.883268 | Actin | AT3G46520 | ACGGGTATCGTGCTTGACTC | 59.83 | 182 | 108.36% | 0.9957 |
| GAACAATTTCCGCTCGGCAG | 60.18 | |||||||
| ACT1 | SRR7873856.1.2050798 | Actin | AT2G37620 | GTCGGGACTGTGTGACTGAC | 60.23 | 193 | 107.67% | 0.9983 |
| CCCGGAAGAGCACCTAACTC | 59.82 | |||||||
| 18S rRNA | SRR7873856.1.1835427 | 18S ribosomal RNA | AT3G41768 | GACGGAGGTAGGGTTCGATT | 58.89 | 197 | 108.49% | 0.9933 |
| CACCAGACTTGCCTCCAATG | 58.83 | |||||||
| eIF-3 | SRR7873856.1.968612 | Translation initiation factor | AT4G20980 | GGCTCTTGAGTCGCTCCAAT | 60.11 | 195 | 107.04% | 0.9993 |
| GCGAATCGTCGGTGTTCAAG | 59.91 | |||||||
| eIF4A-III | SRR7873856.1.1836036 | Translation initiation factor | AT3G19760 | CCACCTTTTGCCTCCAACAC | 59.61 | 191 | 106.06% | 0.9971 |
| GGTACCGGGAAAACCTCCAT | 59.38 | |||||||
| eIF-2 | SRR7873856.1.18172 | Translation initiation factor | AT1G76720 | TTTTGGGAGAGCGGACACAA | 59.82 | 196 | 102.96% | 0.9927 |
| AGCTGCCTTGGAGACTGAAA | 59.23 | |||||||
| His3.3 | SRR7873856.1.2135040 | Histone | AT4G40030 | CACAAGGTAGGCCTCTGCTG | 60.39 | 193 | 105.94% | 0.9945 |
| AAGAAGCCCACAGATACCGC | 60.11 | |||||||
| TUA6 | SRR7873856.1.1877380 | α-tubulin | AT4G14960 | TCCACCCACTCCCTTCTTGA | 60.10 | 193 | 105.31% | 0.9960 |
| TTCATCCACGTTCAGGCTCC | 60.04 | |||||||
| CYP38 | SRR7873856.1.1584051 | Cyclophilin | AT3G01480 | CGCTCGAGAGGGTCGATAAC | 60.04 | 188 | 102.00% | 0.9921 |
| GCCTGCTACCACTTGACTGA | 59.68 | |||||||
| PP2A-2 | SRR7873856.1.2107443 | Protein phosphatase 2Asubunit | AT1G10430 | TTTAGATCAGAGGTGCGCGG | 60.18 | 185 | 106.41% | 0.9945 |
| AAATTGCTCTCGCGCCTGAT | 60.75 | |||||||
| PP2A-3 | SRR7873856.1.2002889 | Protein phosphatase 2A subunit | AT2G42500 | CCAGCACCTCGAGGGAGATA | 60.47 | 182 | 103.48% | 0.9971 |
| TTCCGACTGCACTGGTTGAA | 59.82 | |||||||
| PP2A-4 | SRR7873856.1.256172 | Protein phosphatase 2A subunit | AT3G58500 | GTGGCTTTGAAAGTGCGCTA | 59.41 | 184 | 100.20% | 0.9936 |
| TGATTCAACCAAGGCGGTCA | 59.89 | |||||||
| VAB2 | SRR7873856.1.1180932 | Homeodomain transcription factor | AT3G05020 | CTCGGAATTGTCGTCAGGCT | 60.11 | 197 | 98.37% | 0.9943 |
| ATCGTTGGCCGTTCAGGAAA | 60.25 |
| Rank | Total Samples | Different Organs | Different Periods | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | CV ± SD | r | p-Value | Gene | CV ± SD | r | p-Value | Gene | CV ± SD | r | p-Value | |
| 1 | His3.3 | 1.50 ± 0.33 | 0.644 | 0.005 | His3.3 | 1.42 ± 0.32 | 0.672 | 0.033 | eIF-2 | 1.60 ± 0.34 | 0.910 | 0.001 |
| 2 | eIF-2 | 1.76 ± 0.38 | 0.926 | 0.001 | eIF-2 | 1.79 ± 0.39 | 0.937 | 0.001 | His3.3 | 1.93 ± 0.43 | 0.799 | 0.010 |
| 3 | eIF4A-3 | 2.85 ± 0.66 | 0.663 | 0.004 | eIF4A-3 | 3.05 ± 0.71 | 0.573 | 0.083 | PP2A-2 | 1.95 ± 0.45 | 0.208 | 0.593 |
| 4 | PP2A-2 | 3.30 ± 0.77 | 0.489 | 0.046 | CYP38 | 4.34 ± 1.18 | 0.861 | 0.001 | eIF4A-3 | 2.23 ± 0.51 | 0.836 | 0.005 |
| 5 | ACT1 | 3.32 ± 1.15 | 0.321 | 0.210 | eIF-3 | 4.41 ± 1.08 | 0.306 | 0.389 | ACT7 | 2.56 ± 0.59 | 0.513 | 0.810 |
| 6 | eIF-3 | 3.77 ± 0.91 | 0.028 | 0.914 | ACT1 | 4.46 ± 1.54 | 0.208 | 0.565 | ACT1 | 2.88 ± 1.01 | 0.950 | 0.001 |
| 7 | ACT7 | 3.97 ± 0.91 | 0.527 | 0.030 | PP2A-2 | 4.52 ± 1.07 | 0.810 | 0.004 | ACT12 | 3.24 ± 0.71 | 0.108 | 0.780 |
| 8 | CYP38 | 4.66 ± 1.28 | 0.699 | 0.002 | PP2A-3 | 4.62 ± 1.06 | 0.934 | 0.001 | eIF-3 | 3.48 ± 0.83 | 0.534 | 0.055 |
| 9 | ACT12 | 4.71 ± 1.02 | 0.454 | 0.068 | ACT7 | 4.71 ± 1.07 | 0.864 | 0.001 | PP2A-3 | 4.52 ± 1.12 | 0.924 | 0.001 |
| 10 | PP2A-3 | 4.93 ± 1.17 | 0.907 | 0.001 | TUA6 | 4.99 ± 1.02 | 0.808 | 0.005 | TUA6 | 4.59 ± 0.94 | 0.889 | 0.001 |
| 11 | TUA6 | 4.94 ± 1.01 | 0.837 | 0.001 | ACT12 | 5.79 ± 1.25 | 0.745 | 0.013 | CYP38 | 5.08 ± 1.44 | 0.709 | 0.032 |
| 12 | PP2A-4 | 6.51 ± 1.49 | 0.998 | 0.001 | PP2A-4 | 7.43 ± 1.67 | 0.992 | 0.001 | PP2A-4 | 6.81 ± 1.62 | 0.995 | 0.001 |
| 13 | VAB2 | 7.43 ± 1.68 | 0.995 | 0.001 | VAB2 | 8.99 ± 1.99 | 0.995 | 0.001 | VAB2 | 7.01 ± 1.67 | 0.999 | 0.001 |
| 14 | 18S rRNA | 19.52 ± 1.54 | 0.895 | 0.001 | 18S rRNA | 15.67 ± 1.27 | 0.944 | 0.001 | 18S rRNA | 24.88 ± 2.04 | 0.931 | 0.001 |
| Group | Rank | Delta Ct | BestKeeper | NormFinder | geNorm | RefFinder | |
|---|---|---|---|---|---|---|---|
| Gene | Gene | Gene | Gene | Gene | SV | ||
| Total samples | 1 | His3.3 | His3.3 | eIF-2 | eIF-2 | eIF-2 | 1.190 |
| 2 | eIF-2 | eIF-2 | TUA6 | His3.3 | His3.3 | 1.570 | |
| 3 | eIF4A-3 | eIF4A-3 | His3.3 | TUA6 | PP2A-2 | 3.940 | |
| 4 | PP2A-2 | PP2A-2 | eIF4A-3 | PP2A-2 | TUA6 | 3.980 | |
| 5 | ACT7 | ACT1 | PP2A-2 | eIF4A-3 | eIF4A-3 | 4.530 | |
| 6 | TUA6 | eIF-3 | ACT7 | ACT7 | ACT7 | 5.180 | |
| 7 | ACT12 | ACT7 | ACT12 | ACT12 | ACT12 | 6.650 | |
| 8 | eIF-3 | CYP38 | PP2A-3 | PP2A-3 | PP2A-3 | 8.460 | |
| 9 | ACT1 | ACT12 | CYP38 | PP2A-4 | PP2A-4 | 9.930 | |
| 10 | PP2A-3 | PP2A-3 | PP2A-4 | CYP38 | CYP38 | 9.970 | |
| 11 | CYP38 | TUA6 | 18S rRNA | 18S rRNA | eIF-3 | 10.920 | |
| 12 | 18S rRNA | PP2A-4 | VAB2 | VAB2 | 18S rRNA | 11.470 | |
| 13 | PP2A-4 | VAB2 | ACT1 | eIF-3 | ACT1 | 12.310 | |
| 14 | VAB2 | 18S rRNA | eIF-3 | ACT1 | VAB2 | 12.470 | |
| Different organs | 1 | eIF-2 | His3.3 | TUA6 | TUA6 | TUA6 | 1.410 |
| 2 | His3.3 | eIF-2 | PP2A-3 | PP2A-3 | PP2A-2 | 3.220 | |
| 3 | PP2A-2 | eIF4A-3 | eIF-2 | PP2A-2 | eIF-2 | 3.310 | |
| 4 | eIF4A-3 | CYP38 | ACT7 | eIF-2 | PP2A-3 | 3.440 | |
| 5 | ACT7 | eIF-3 | His3.3 | ACT7 | His3.3 | 3.810 | |
| 6 | ACT12 | ACT1 | PP2A-2 | ACT12 | ACT7 | 4.860 | |
| 7 | eIF-3 | PP2A-2 | ACT12 | His3.3 | ACT12 | 5.960 | |
| 8 | TUA6 | PP2A-3 | 18S rRNA | 18S rRNA | eIF4A-3 | 7.000 | |
| 9 | ACT1 | ACT7 | CYP38 | CYP38 | 18S rRNA | 8.920 | |
| 10 | PP2A-3 | TUA6 | eIF4A-3 | eIF4A-3 | CYP38 | 9.240 | |
| 11 | CYP38 | ACT12 | PP2A-4 | PP2A-4 | eIF-3 | 10.610 | |
| 12 | PP2A-4 | PP2A-4 | eIF-3 | eIF-3 | PP2A-4 | 11.720 | |
| 13 | VAB2 | VAB2 | VAB2 | VAB2 | VAB2 | 13.240 | |
| 14 | 18S rRNA | 18S rRNA | ACT1 | ACT1 | ACT1 | 13.470 | |
| Different periods | 1 | His3.3 | eIF-2 | eIF4A-3 | eIF-2 | eIF-2 | 1.190 |
| 2 | eIF-2 | His3.3 | eIF-2 | His3.3 | His3.3 | 1.860 | |
| 3 | eIF4A-3 | PP2A-2 | His3.3 | eIF4A-3 | eIF4A-3 | 2.450 | |
| 4 | TUA6 | eIF4A-3 | TUA6 | TUA6 | PP2A-2 | 4.560 | |
| 5 | PP2A-2 | ACT7 | ACT1 | ACT1 | TUA6 | 5.470 | |
| 6 | ACT7 | ACT1 | PP2A-2 | PP2A-2 | ACT1 | 6.510 | |
| 7 | eIF-3 | ACT12 | PP2A-3 | PP2A-3 | ACT7 | 7.580 | |
| 8 | ACT12 | eIF-3 | PP2A-4 | PP2A-4 | ACT12 | 7.750 | |
| 9 | PP2A-3 | PP2A-3 | VAB2 | VAB2 | PP2A-3 | 8.150 | |
| 10 | CYP38 | TUA6 | ACT12 | ACT12 | PP2A-4 | 9.360 | |
| 11 | 18S rRNA | CYP38 | CYP38 | ACT7 | VAB2 | 10.370 | |
| 12 | ACT1 | PP2A-4 | ACT7 | CYP38 | CYP38 | 11.490 | |
| 13 | PP2A-4 | VAB2 | 18S rRNA | 18S rRNA | eIF-3 | 11.770 | |
| 14 | VAB2 | 18S rRNA | eIF-3 | eIF-3 | 18S rRNA | 13.240 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Zhao, H.; Zhang, X.; Liang, Z.; He, Q. Systematic Identification and Validation of Suitable Reference Genes for the Normalization of Gene Expression in Prunella vulgaris under Different Organs and Spike Development Stages. Genes 2022, 13, 1947. https://doi.org/10.3390/genes13111947
Zheng H, Zhao H, Zhang X, Liang Z, He Q. Systematic Identification and Validation of Suitable Reference Genes for the Normalization of Gene Expression in Prunella vulgaris under Different Organs and Spike Development Stages. Genes. 2022; 13(11):1947. https://doi.org/10.3390/genes13111947
Chicago/Turabian StyleZheng, Hui, Hongguang Zhao, Xuemin Zhang, Zongsuo Liang, and Qiuling He. 2022. "Systematic Identification and Validation of Suitable Reference Genes for the Normalization of Gene Expression in Prunella vulgaris under Different Organs and Spike Development Stages" Genes 13, no. 11: 1947. https://doi.org/10.3390/genes13111947
APA StyleZheng, H., Zhao, H., Zhang, X., Liang, Z., & He, Q. (2022). Systematic Identification and Validation of Suitable Reference Genes for the Normalization of Gene Expression in Prunella vulgaris under Different Organs and Spike Development Stages. Genes, 13(11), 1947. https://doi.org/10.3390/genes13111947