
Reclassification of DMD Duplications as Benign: Recommendations for Cautious Interpretation of Variants Identified in Prenatal Screening

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
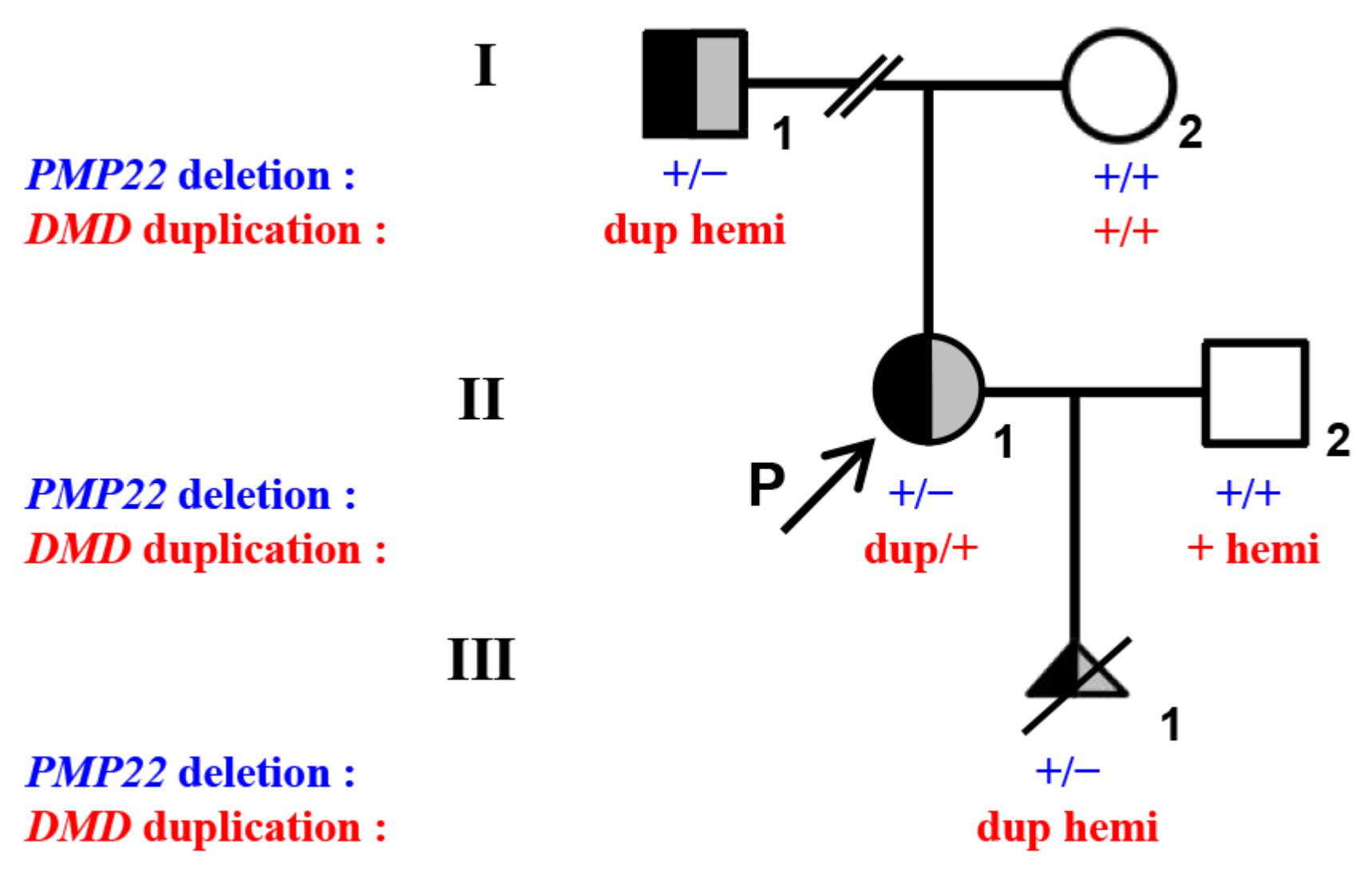
2.1. Patients
2.2. MLPA and Quantitative Polymerase Chain Reaction (qPCR)
2.3. DMD Sequencing and Analysis by Bionano SaphyrTM
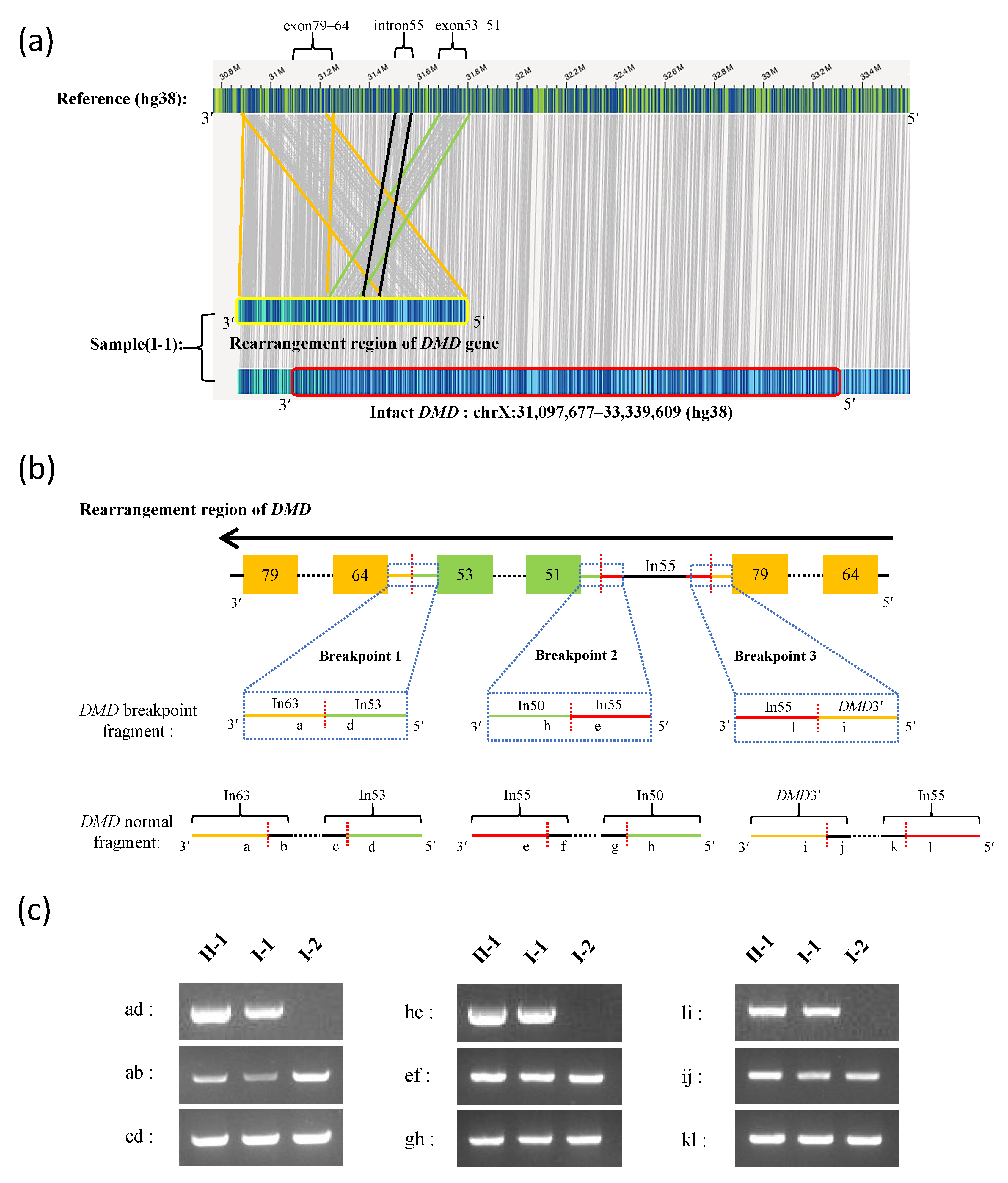
2.4. Breakpoint Analysis and Sanger Sequencing
3. Results
3.1. MLPA and q-PCR Analysis
3.2. Long-Read Sequencing and Breakpoint Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flanigan, K.M. Duchenne and Becker Muscular Dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population Frequencies of Inherited Neuromuscular Diseases—A World Survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-Based Path to Newborn Screening for Duchenne Muscular Dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Happi Mbakam, C.; Lamothe, G.; Tremblay, J.P. Therapeutic Strategies for Dystrophin Replacement in Duchenne Muscular Dystrophy. Front. Med. 2022, 9, 859930. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Grootscholten, P.M.; Dauwerse, J.G.; Walker, A.P.; Monaco, A.P.; Butler, R.; Anand, R.; Coffey, A.J.; Bentley, D.R.; Steensma, H.Y. Reconstruction of the 2.4 Mb Human DMD-Gene by Homologous YAC Recombination. Hum. Mol. Genet. 1992, 1, 19–28. [Google Scholar] [CrossRef]
- Kong, X.; Zhong, X.; Liu, L.; Cui, S.; Yang, Y.; Kong, L. Genetic Analysis of 1051 Chinese Families with Duchenne/Becker Muscular Dystrophy. BMC Med. Genet. 2019, 20, 139. [Google Scholar] [CrossRef] [Green Version]
- Zamani, G.; Hosseinpour, S.; Ashrafi, M.R.; Mohammadi, M.; Badv, R.S.; Tavasoli, A.R.; Akbari, M.G.; Bereshneh, A.H.; Malamiri, R.A.; Heidari, M. Characteristics of Disease Progression and Genetic Correlation in Ambulatory Iranian Boys with Duchenne Muscular Dystrophy. BMC Neurol. 2022, 22, 162. [Google Scholar] [CrossRef]
- Brison, N.; Storms, J.; Villela, D.; Claeys, K.G.; Dehaspe, L.; de Ravel, T.; De Waele, L.; Goemans, N.; Legius, E.; Peeters, H.; et al. Maternal Copy-Number Variations in the DMD Gene as Secondary Findings in Noninvasive Prenatal Screening. Genet. Med. 2019, 21, 2774–2780. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Kroon, M.; López Hernández, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 Platform: Efficient Genome-Wide Sharing of Genetic Variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef]
- Goldrich, D.Y.; LaBarge, B.; Chartrand, S.; Zhang, L.; Sadowski, H.B.; Zhang, Y.; Pham, K.; Way, H.; Lai, C.-Y.J.; Pang, A.W.C.; et al. Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping. J. Pers. Med. 2021, 11, 142. [Google Scholar] [CrossRef]
- Stence, A.A.; Thomason, J.G.; Pruessner, J.A.; Sompallae, R.R.; Snow, A.N.; Ma, D.; Moore, S.A.; Bossler, A.D. Validation of Optical Genome Mapping for the Molecular Diagnosis of Facioscapulohumeral Muscular Dystrophy. J. Mol. Diagn. 2021, 23, 1506–1514. [Google Scholar] [CrossRef]
- Barseghyan, H.; Tang, W.; Wang, R.T.; Almalvez, M.; Segura, E.; Bramble, M.S.; Lipson, A.; Douine, E.D.; Lee, H.; Délot, E.C.; et al. Next-Generation Mapping: A Novel Approach for Detection of Pathogenic Structural Variants with a Potential Utility in Clinical Diagnosis. Genome Med. 2017, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Carsana, A.; Frisso, G.; Intrieri, M.; Tremolaterra, M.R.; Savarese, G.; Scapagnini, G.; Esposito, G.; Santoro, L.; Salvatore, F. A 15-Year Molecular Analysis of DMD/BMD: Genetic Features in a Large Cohort. Front. Biosci. 2010, 2, 547–558. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; den Dunnen, J.T. Phenotype Predictions for Exon Deletions/Duplications: A User Guide for Professionals and Clinicians Using Becker and Duchenne Muscular Dystrophy as Examples. Hum. Mutat. 2019, 40, 1630–1633. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, J.; Xu, J.; Sun, Y.; Li, J.; Gao, Y.; Liu, L.; Jia, C.; Kong, X.; Wang, L. Long-Read Sequencing Revealed Extragenic and Intragenic Duplications of Exons 56-61 in DMD in an Asymptomatic Male and a DMD Patient. Front. Genet. 2022, 13, 878806. [Google Scholar] [CrossRef]
- White, S.J.; Aartsma-Rus, A.; Flanigan, K.M.; Weiss, R.B.; Kneppers, A.L.J.; Lalic, T.; Janson, A.a.M.; Ginjaar, H.B.; Breuning, M.H.; den Dunnen, J.T. Duplications in the DMD Gene. Hum. Mutat. 2006, 27, 938–945. [Google Scholar] [CrossRef]
- Baskin, B.; Stavropoulos, D.J.; Rebeiro, P.A.; Orr, J.; Li, M.; Steele, L.; Marshall, C.R.; Lemire, E.G.; Boycott, K.M.; Gibson, W.; et al. Complex Genomic Rearrangements in the Dystrophin Gene Due to Replication-Based Mechanisms. Mol. Genet. Genom. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Luce, L.; Abelleyro, M.M.; Carcione, M.; Mazzanti, C.; Rossetti, L.; Radic, P.; Szijan, I.; Menazzi, S.; Francipane, L.; Nevado, J.; et al. Analysis of Complex Structural Variants in the DMD Gene in One Family. Neuromuscul. Disord. 2021, 31, 253–263. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Chen, J.-M.; Cooper, D.N.; Férec, C.; Kehrer-Sawatzki, H.; Patrinos, G.P. Genomic Rearrangements in Inherited Disease and Cancer. Semin. Cancer Biol. 2010, 20, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Dai, Y.; Fang, L.; Yao, F.; Liu, Z.; Qiu, Z.; Cui, L.; Xia, F.; Zhao, C.; Zhang, S.; et al. Exonic Rearrangements in DMD in Chinese Han Individuals Affected with Duchenne and Becker Muscular Dystrophies. Hum. Mutat. 2020, 41, 668–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishmukhametova, A.; Khau Van Kien, P.; Méchin, D.; Thorel, D.; Vincent, M.-C.; Rivier, F.; Coubes, C.; Humbertclaude, V.; Claustres, M.; Tuffery-Giraud, S. Comprehensive Oligonucleotide Array-Comparative Genomic Hybridization Analysis: New Insights into the Molecular Pathology of the DMD Gene. Eur. J. Hum. Genet. 2012, 20, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Maya, I.; Salzer Sheelo, L.; Brabbing-Goldstein, D.; Matar, R.; Kahana, S.; Agmon-Fishman, I.; Klein, C.; Gurevitch, M.; Basel-Salmon, L.; Sagi-Dain, L. Residual Risk for Clinically Significant Copy Number Variants in Low-Risk Pregnancies, Following Exclusion of Noninvasive Prenatal Screening-Detectable Findings. Am. J. Obstet. Gynecol. 2022, 226, 562.e1–562.e8. [Google Scholar] [CrossRef]
- Advani, H.V.; Barrett, A.N.; Evans, M.I.; Choolani, M. Challenges in Non-Invasive Prenatal Screening for Sub-Chromosomal Copy Number Variations Using Cell-Free DNA. Prenat. Diagn. 2017, 37, 1067–1075. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Bejjani, B.A.; Torchia, B.; Kirkpatrick, S.; Coppinger, J.; Ballif, B.C. The Identification of Microdeletion Syndromes and Other Chromosome Abnormalities: Cytogenetic Methods of the Past, New Technologies for the Future. Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145C, 335–345. [Google Scholar] [CrossRef]
- Van Paassen, B.W.; van der Kooi, A.J.; van Spaendonck-Zwarts, K.Y.; Verhamme, C.; Baas, F.; de Visser, M. PMP22 Related Neuropathies: Charcot-Marie-Tooth Disease Type 1A and Hereditary Neuropathy with Liability to Pressure Palsies. Orphanet. J. Rare Dis. 2014, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Casasnovas, C.; Banchs, I.; De Jorge, L.; Antónia Albertí, M.; Martínez-Campo, Y.; Povedano, M.; Montero, J.; Volpini, V. A Novel Small Deletion in PMP22 Causes a Mild Hereditary Neuropathy with Liability to Pressure Palsies Phenotype. Muscle Nerve 2012, 45, 135–138. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Breakpoint Junction Sequence (5′-3′) | Breakpoint 5′ Region | Breakpoint 5′ Sequence | Breakpoint 5′ Site | Breakpoint 3′ Region | Breakpoint 3′ Sequence | Breakpoint 3′ Site |
|---|---|---|---|---|---|---|---|
| Breakpoint 1 | TAATGAAAAG|GTTTTGTTTT | Intron 53 | TAATGAAAAG | ChrX: 31,674,049 | Intron 63 | GTTTTGTTTT | ChrX: 31,260,251 |
| Breakpoint 2 | CAACAACAGA|CATTTAGATG | Intron 55 | CAACAACAGA | ChrX: 31,513,138 | Intron 50 | CATTTAGATG | ChrX: 31,811,202 |
| Breakpoint 3 | AATATTTTTA|TCTTTGAAGC | DMD3′ | AATATTTTTA | ChrX: 30,880,778 | Intron 55 | TCTTTGAAGC | ChrX: 31,570,889 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, W.; Meng, G.; Hu, X.; Dai, J.; Liu, J.; Li, X.; Hu, H.; Tan, Y.; Zhang, Q.; Lu, G.; et al. Reclassification of DMD Duplications as Benign: Recommendations for Cautious Interpretation of Variants Identified in Prenatal Screening. Genes 2022, 13, 1972. https://doi.org/10.3390/genes13111972
He W, Meng G, Hu X, Dai J, Liu J, Li X, Hu H, Tan Y, Zhang Q, Lu G, et al. Reclassification of DMD Duplications as Benign: Recommendations for Cautious Interpretation of Variants Identified in Prenatal Screening. Genes. 2022; 13(11):1972. https://doi.org/10.3390/genes13111972
Chicago/Turabian StyleHe, Wenbin, Guiquan Meng, Xiao Hu, Jing Dai, Jiyang Liu, Xiurong Li, Hao Hu, Yueqiu Tan, Qianjun Zhang, Guangxiu Lu, and et al. 2022. "Reclassification of DMD Duplications as Benign: Recommendations for Cautious Interpretation of Variants Identified in Prenatal Screening" Genes 13, no. 11: 1972. https://doi.org/10.3390/genes13111972
APA StyleHe, W., Meng, G., Hu, X., Dai, J., Liu, J., Li, X., Hu, H., Tan, Y., Zhang, Q., Lu, G., Lin, G., & Du, J. (2022). Reclassification of DMD Duplications as Benign: Recommendations for Cautious Interpretation of Variants Identified in Prenatal Screening. Genes, 13(11), 1972. https://doi.org/10.3390/genes13111972