
Genome-Wide Association Study of Growth Traits in a Four-Way Crossbred Pig Population
Abstract
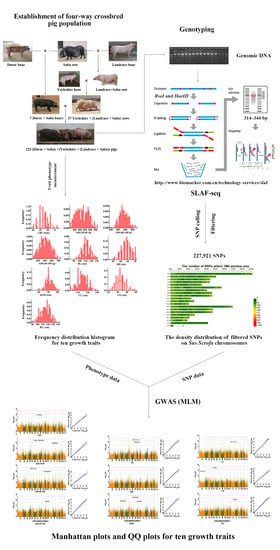
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals, Phenotypic Collection and Statistical Analysis
2.3. SLAF Library Construction and High-Throughput Sequencing
2.4. Data Processing and SNP Calling
2.5. Genome-Wide Association Study (GWAS)
2.6. Identification and Annotation of Candidate Genes
2.7. Association Analysis between SNP Marker Genotypes and Growth Traits
3. Results
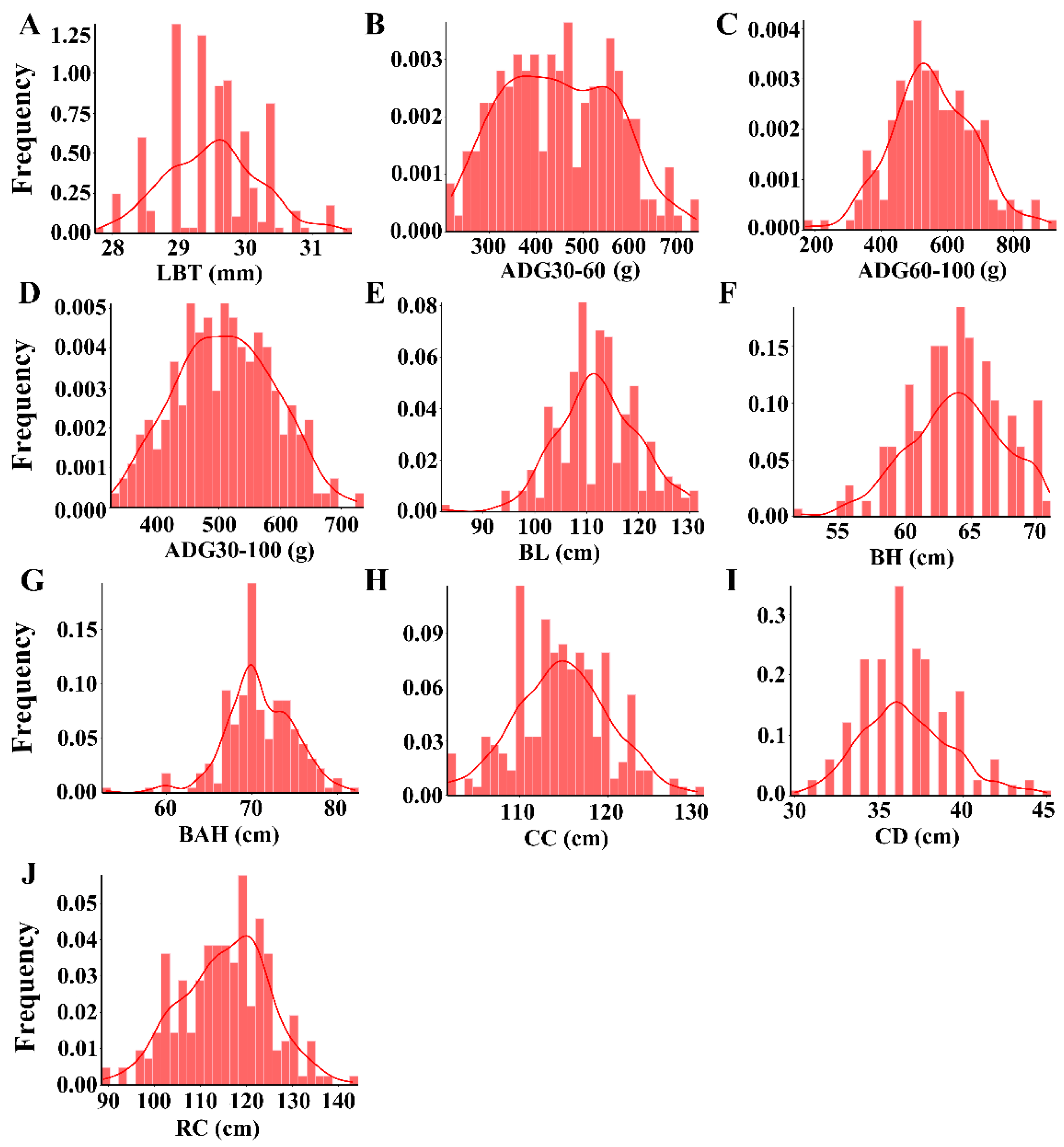
3.1. Phenotype Description and Correlation among Growth Traits
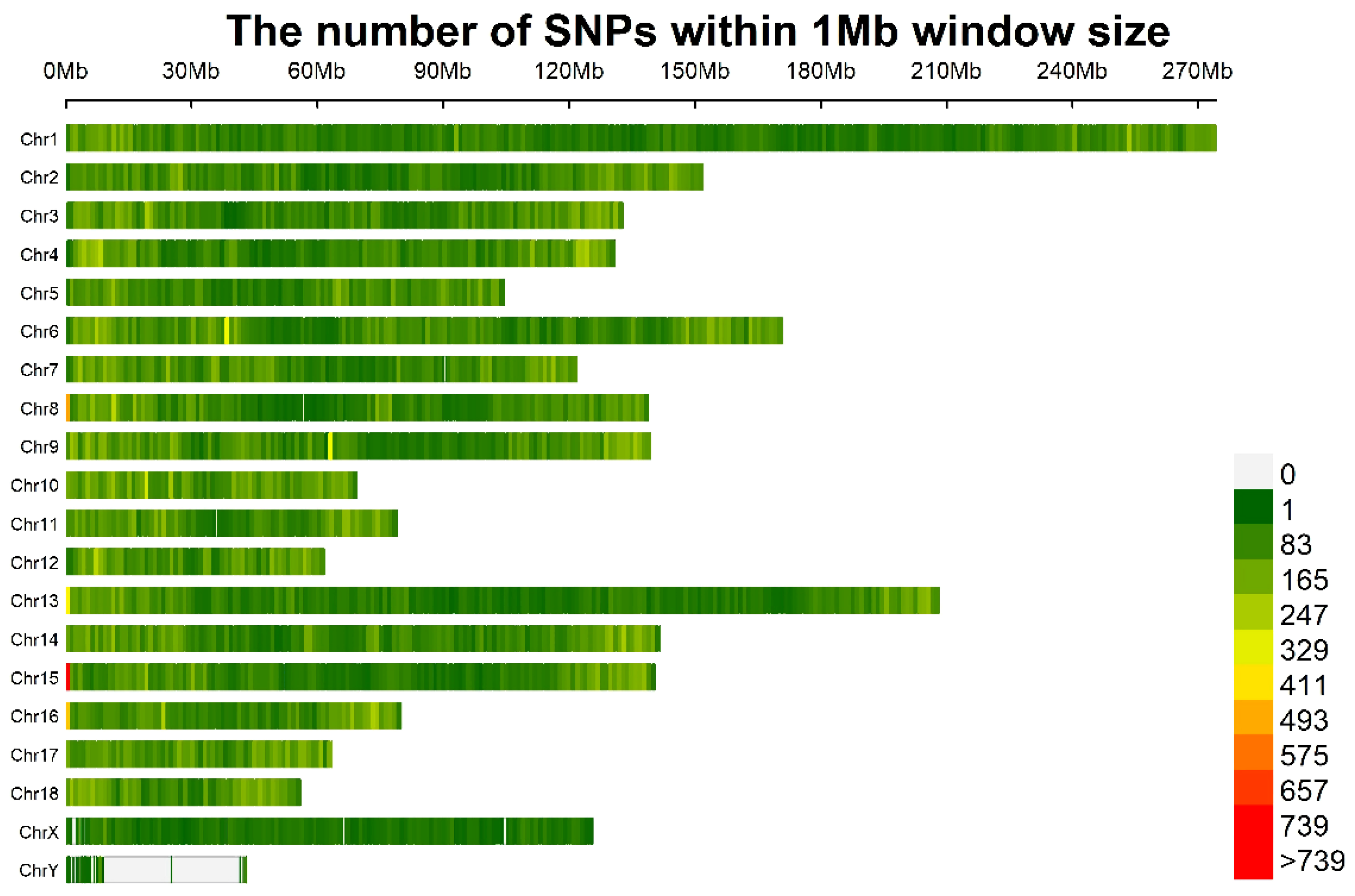
3.2. Identification of SLAFs and SNPs
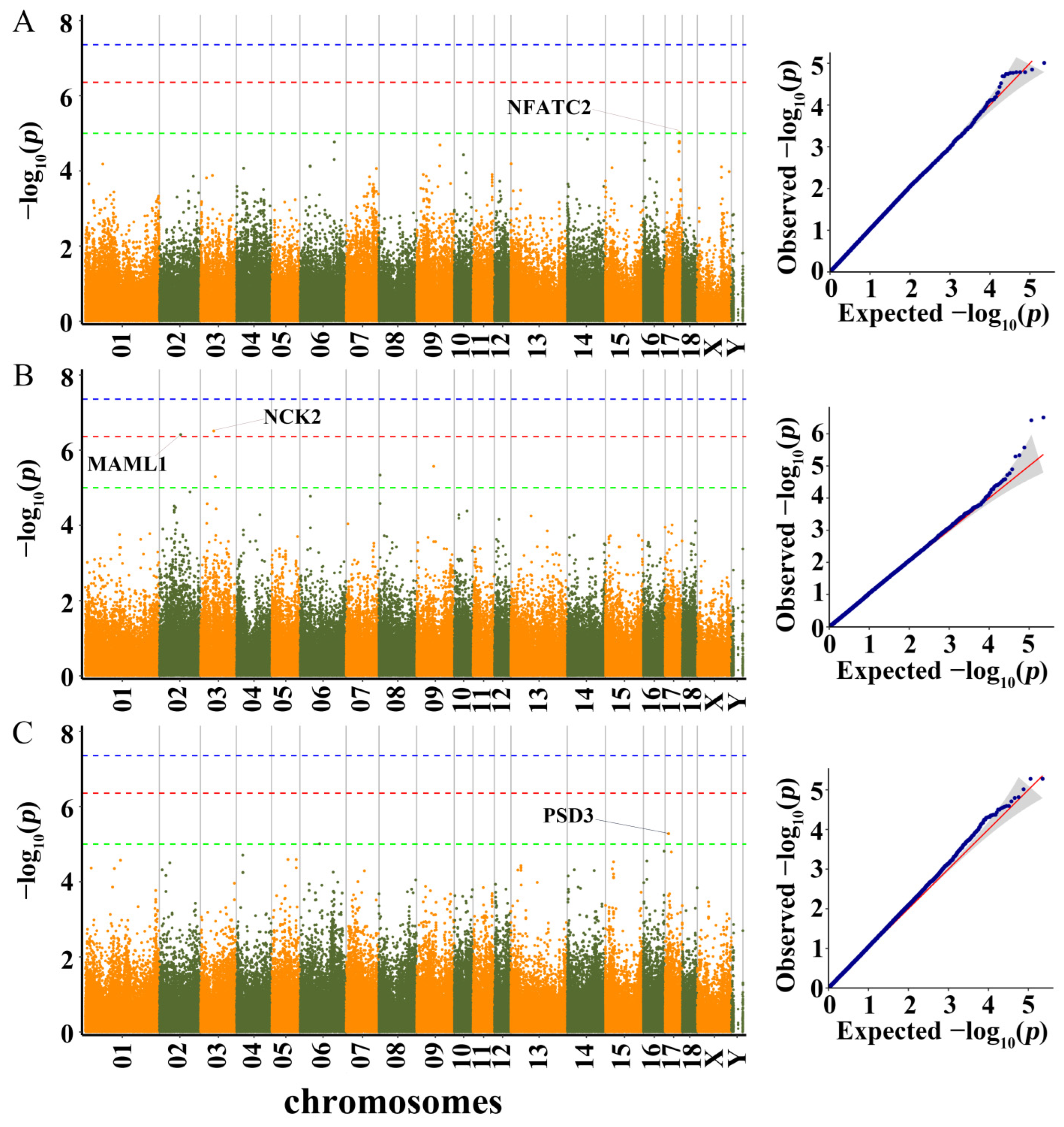
3.3. Genome-Wide Association Study and Identification of Candidate Genes
3.3.1. LBT, ADG30–60, ADG60–100, and ADG30–100
3.3.2. BL, BH, and BAH
3.3.3. CC, CD, and RC
3.4. Comparison with Previously Mapped QTL in Pigs
3.5. Functional Annotation of Candidate Genes
3.6. Association Analysis between SNP Marker Genotypes and Growth Traits
4. Discussion
4.1. QTLs Identified for Growth Traits
4.2. Candidate Genes for LBT, ADG30–60, ADG60–100 and ADG30–100
4.3. Candidate Genes for BL and BAH
4.4. Candidate Genes for CC, CD and RC
4.5. Comparison between ADG30–60 and ADG60–100
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.; Russo, V. A Genomewide Association Study for Average Daily Gain in Italian Large White Pigs. J. Anim. Sci. 2014, 92, 1385–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhuang, Z.; Yang, M.; Ding, R.; Quan, J.; Zhou, S.; Gu, T.; Xu, Z.; Zheng, E.; Cai, G.; et al. Genome-Wide Detection of Genetic Loci and Candidate Genes for Body Conformation Traits in Duroc × Landrace × Yorkshire Crossbred Pigs. Front. Genet. 2021, 12, 664343. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Song, H.; Jiang, Y.; Jiang, Y.; Zhang, F.; Liu, Y.; Shi, Y.; Ding, X.; Wang, C. A Single-Step Genome Wide Association Study on Body Size Traits Using Imputation-Based Whole-Genome Sequence Data in Yorkshire Pigs. Front. Genet. 2021, 12, 629049. [Google Scholar] [CrossRef]
- Wang, Y.; Thakali, K.; Morse, P.; Shelby, S.; Chen, J.; Apple, J.; Huang, Y. Comparison of Growth Performance and Meat Quality Traits of Commercial Cross-Bred Pigs versus the Large Black Pig Breed. Animals 2021, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Carcò, G.; Gallo, L.; Dalla Bona, M.; Latorre, M.A.; Fondevila, M.; Schiavon, S. The Influence of Feeding Behaviour on Growth Performance, Carcass and Meat Characteristics of Growing Pigs. PLoS ONE 2018, 13, e0205572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.W.; Yan, L.; Ao, X.; Zhou, T.X.; Wang, J.P.; Lee, J.H.; Kim, I.H. Influence of Probiotics in Different Energy and Nutrient Density Diets on Growth Performance, Nutrient Digestibility, Meat Quality, and Blood Characteristics in Growing-Finishing Pigs. J. Anim. Sci. 2010, 88, 3320–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, L. Genome-Wide Association Analysis in Domestic Animals: A Powerful Approach for Genetic Dissection of Trait Loci. Genetica 2009, 136, 341–349. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, K.; Liu, X.; Zhou, H.; Xu, L.; Wang, Z.; Fang, M. Identification of Growth Trait Related Genes in a Yorkshire Purebred Pig Population by Genome-Wide Association Studies. Asian-Aust. J. Anim. Sci. 2017, 30, 462–469. [Google Scholar] [CrossRef]
- Tang, Z.; Xu, J.; Yin, L.; Yin, D.; Zhu, M.; Yu, M.; Li, X.; Zhao, S.; Liu, X. Genome-Wide Association Study Reveals Candidate Genes for Growth Relevant Traits in Pigs. Front. Genet. 2019, 10, 302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, Z.T.; Diao, S.Q.; Ye, S.P.; Wang, J.Y.; Gao, N.; Yuan, X.L.; Chen, Z.M.; Zhang, H.; Li, J.Q. Identifying the Complex Genetic Architecture of Growth and Fatness Traits in a Duroc Pig Population. J. Integr. Agr. 2021, 20, 1607–1614. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, R.; Meng, F.; Wang, X.; Zhuang, Z.; Quan, J.; Geng, Q.; Wu, J.; Zheng, E.; Wu, Z.; et al. A Meta-Analysis of Genome-Wide Association Studies for Average Daily Gain and Lean Meat Percentage in Two Duroc Pig Populations. BMC Genom. 2021, 22, 12. [Google Scholar] [CrossRef]
- Qiao, R.; Gao, J.; Zhang, Z.; Li, L.; Xie, X.; Fan, Y.; Cui, L.; Ma, J.; Ai, H.; Ren, J.; et al. Genome-Wide Association Analyses Reveal Significant Loci and Strong Candidate Genes for Growth and Fatness Traits in Two Pig Populations. Genet. Sel. Evol. 2015, 47, 17. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Xiao, S.; Li, W.; Huang, T.; Huang, X.; Yan, G.; Huang, Y.; Qiu, H.; Jiang, K.; Wang, X.; et al. Unravelling the Genetic Loci for Growth and Carcass Traits in Chinese Bamaxiang Pigs Based on a 1.4 Million SNP Array. J. Anim. Breed. Genet. 2019, 136, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tang, S.; Wang, C.; Wang, Y.; Qin, Y.; Wang, Y.; Zhang, J.; Song, H.; Mi, S.; Yu, F.; et al. A Genome-Wide Association Study of Growth and Fatness Traits in Two Pig Populations with Different Genetic Backgrounds. J. Anim. Sci. 2018, 96, 806–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.H.; Christensen, O.F.; Nielsen, B.; Sahana, G. Genome-Wide Association Study for Conformation Traits in Three Danish Pig Breeds. Genet. Sel. Evol. 2017, 49, 12. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zhou, L.; Guo, Y.; Huang, L.; Ma, J. Genome-Wide Association Study Identifies 22 New Loci for Body Dimension and Body Weight Traits in a White Duroc × Erhualian F2 Intercross Population. Asian-Australas J. Anim. Sci. 2017, 30, 1066–1073. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Liu, S.; Li, W.; Mao, R.; Zhuo, Y.; Xing, W.; Liu, J.; Wang, C.; Zhou, L.; Lei, M.; et al. Genome-Wide Association Study Reveals Additive and Non-Additive Effects on Growth Traits in Duroc Pigs. Genes 2022, 13, 1454. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, Y.; Hou, L.; Ma, J.; Chen, C.; Ai, H.; Huang, L.; Ren, J. Genome-Wide Detection of Genetic Markers Associated with Growth and Fatness in Four Pig Populations using Four Approaches. Genet. Sel. Evol. 2017, 49, 21. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Wu, J.; Yu, J.; Zheng, X.; Kang, H.; Wang, Z.; Zhang, S.; Zhou, L.; Liu, J. A Genome-Wide Association Study Reveals Additive and Dominance Effects on Growth and Fatness Traits in Large White Pigs. Anim. Genet. 2021, 52, 749–753. [Google Scholar] [CrossRef]
- Falker-Gieske, C.; Blaj, I.; Preuss, S.; Bennewitz, J.; Thaller, G.; Tetens, J. GWAS for Meat and Carcass Traits Using Imputed Sequence Level Genotypes in Pooled F2-Designs in Pigs. G3 Genes Genom. Genet. 2019, 9, 2823–2834. [Google Scholar] [CrossRef]
- Blaj, I.; Tetens, J.; Preuss, S.; Bennewitz, J.; Thaller, G. Genome-Wide Association Studies and Meta-Analysis Uncovers New Candidate Genes for Growth and Carcass Traits in Pigs. PLoS ONE 2018, 13, e0205576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, D.; Zhuang, Z.; Ding, R.; Qiu, Y.; Zhou, S.; Wu, J.; Xu, C.; Hong, L.; Huang, S.; Zheng, E.; et al. Weighted Single-Step GWAS Identified Candidate Genes Associated with Growth Traits in A Duroc Pig Population. Genes 2021, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Ruan, G.R.; Su, Y.; Xiao, S.J.; Zhang, Z.Y.; Ren, J.; Ding, N.S.; Huang, L.S. Genome-Wide Association Study Identifies QTLs for EBV of Backfat Thickness and Average Daily Gain in Duroc Pigs. Genetika 2015, 51, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Christensen, O.F.; Lund, M.S.; Ostersen, T.; Sahana, G. Large-Scale Association Study on Daily Weight Gain in Pigs Reveals Overlap of Genetic Factors for Growth in Humans. BMC Genom. 2022, 23, 133. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-Seq: An Efficient Method of Large-Scale De Novo SNP Discovery and Genotyping Using High-Throughput Sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, T.; Zhang, G.; Wang, J.; Han, K.; Wang, Y.; Zhang, Y. Genome-Wide Association Study of Antibody Level Response to NDV and IBV in Jinghai Yellow Chicken Based on SLAF-Seq Technology. J. Appl. Genet. 2015, 56, 365–373. [Google Scholar] [CrossRef]
- Wang, W.H.; Wang, J.Y.; Zhang, T.; Wang, Y.; Zhang, Y.; Han, K. Genome-Wide Association Study of Growth Traits in Jinghai Yellow Chicken Hens Using SLAF-Seq Technology. Anim. Genet. 2019, 50, 175–176. [Google Scholar] [CrossRef]
- Yang, X.; Deng, F.; Wu, Z.; Chen, S.Y.; Shi, Y.; Jia, X.; Hu, S.; Wang, J.; Cao, W.; Lai, S.J. A Genome-Wide Association Study Identifying Genetic Variants Associated with Growth, Carcass and Meat Quality Traits in Rabbits. Animals 2020, 10, 1068. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.; Liu, W.; Gao, J.; Lei, Q.; Han, H.; Yang, J.; Li, H.; Cao, D.; Zhou, Y. Genome-Wide Association Study of Body Size Traits in Wenshang Barred Chickens Based on the Specific-Locus Amplified Fragment Sequencing Technology. J. Anim. Sci. 2021, 92, e13506. [Google Scholar] [CrossRef]
- Melak, S.; Wang, Q.; Tian, Y.; Wei, W.; Zhang, L.; Elbeltagy, A.; Chen, J. Identification and Validation of Marketing Weight-Related SNP Markers Using SLAF Sequencing in Male Yangzhou Geese. Genes 2021, 12, 1203. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, Q.; Huang, Q.; Ma, S.; Wang, Y.; Han, C.; Zhang, R.; Wang, J.; Liu, H.; Li, L. Genome-Wide Association Analysis Reveals That EDNRB2 Causes a Dose-Dependent Loss of Pigmentation in Ducks. BMC Genom. 2021, 22, 381. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wei, S.; Li, H.; Wu, K.; Cai, Z.; Li, D.; Wei, W.; Li, Q.; Chen, J.; Liu, H.; et al. Genome-Wide Genetic Structure and Differentially Selected Regions among Landrace, Erhualian, and Meishan Pigs Using Specific-Locus Amplified Fragment Sequencing. Sci. Rep. 2017, 7, 10063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, M.; Li, C.; Li, Z.; Chen, W.; Zeng, Y. Genetic Diversities and Differentially Selected Regions between Shandong Indigenous Pig Breeds and Western Pig Breeds. Front. Genet. 2020, 10, 1351. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Yan, D.; Sun, H.; Chen, Q.; Li, M.; Dong, X.; Pan, Y.; Lu, S. Genome-Wide Association Study Identifying Genetic Variants Associated with Carcass Backfat Thickness, Lean Percentage and Fat Percentage in A Four-Way Crossbred Pig Population Using SLAF-Seq Technology. BMC Genom. 2022, 23, 594. [Google Scholar] [CrossRef] [PubMed]
- Diao, S.; Huang, S.; Chen, Z.; Teng, J.; Ma, Y.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs. Genes 2019, 10, 346. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Yang, J.; Goddard, M.E.; Visscher, P.M.; Wray, N.R. Estimation of Pleiotropy between Complex Diseases Using SNP-Derived Genomic Relationships and Restricted Maximum Likelihood. Bioinformatics 2012, 28, 2540–2542. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of A Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the Miseq Illumina Sequencing Platform. Appl. Environ. Microb. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Stephens, M. Genome-Wide Efficient Mixed-Model Analysis for Association Studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-Wide Complex Trait Analysis. Am. J. Hum. Genet 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.J.; Gao, X.F.; Yang, J.; Kong, X.R. Genome-Wide Association Study to Identify Favorable SNP Allelic Variations and Candidate Genes That Control the Timing of Spring Bud Flush of Tea (Camellia Sinensis) Using SLAF-Seq. J. Agric. Food Chem. 2019, 67, 10380–10391. [Google Scholar] [CrossRef]
- Yin, T.; Halli, K.; König, S. Direct Genetic Effects, Maternal Genetic Effects, and Maternal Genetic Sensitivity on Prenatal Heat Stress for Calf Diseases and Corresponding Genomic Loci in German Holsteins. J. Dairy. Sci. 2022, 105, 6795–6808. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Yan, H.; Liu, X.; Li, N.; Liang, J.; Pu, L.; Zhang, Y.; Shi, H.; Zhao, K.; et al. Genome-Wide Association Studies Identify the Loci for 5 Exterior Traits in A Large White × Minzhu Pig Population. PLoS ONE 2014, 9, e103766. [Google Scholar] [CrossRef]
- Rönn, T.; Poulsen, P.; Tuomi, T.; Isomaa, B.; Groop, L.; Vaag, A.; Ling, C. Genetic Variation in ATP5O Is Associated with Skeletal Muscle ATP5O mRNA Expression and Glucose Uptake in Young Twins. PLoS ONE 2009, 4, e4793. [Google Scholar] [CrossRef]
- Giustina, A.; Veldhuis, J.D. Pathophysiology of the Neuroregulation of Growth Hormone Secretion in Experimental Animals and the Human. Endocr. Rev. 1998, 19, 717–797. [Google Scholar] [CrossRef]
- Frohman, L.A.; Kineman, R.D. Growth Hormone-Releasing Hormone and P Development, Hyperplasia and Tumorigenesis. Trends Endocrinol. Metab. 2002, 13, 299–303. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Wang, C.Y.; Kwok, A.H.; Leung, F.C. Identification of the Endogenous Ligands for Chicken Growth Hormone-Releasing Hormone (GHRH) Receptor: Evidence for A Separate Gene Encoding GHRH in Submammalian Vertebrates. Endocrinology 2007, 148, 2405–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoque, M.; Suzuki, K.; Kadowaki, H.; Shibata, T.; Oikawa, T. Genetic Parameters for Feed Efficiency Traits and Their Relationships with Growth and Carcass Traits in Duroc Pigs. J. Anim. Breed. Genet. 2007, 124, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Mavalli, M.D.; DiGirolamo, D.J.; Fan, Y.; Riddle, R.C.; Campbell, K.S.; Van Groen, T.; Frank, S.J.; Sperling, M.A.; Esser, K.A.; Bamman, M.M. Distinct Growth Hormone Receptor Signaling Modes Regulate Skeletal Muscle Development and Insulin Sensitivity in Mice. J. Clin. Investig. 2010, 120, 4007–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Ge, X. Meat Science and Muscle Biology Symposium-Mechanism of Growth Hormone Stimulation of Skeletal Muscle Growth in Cattle. J. Anim. Sci. 2014, 92, 21–29. [Google Scholar] [CrossRef]
- Etherton, T.D.; Wiggins, J.P.; Chung, C.S.; Evock, C.M.; Rebhun, J.F.; Walton, P.E. Stimulation of Pig Growth Performance by Porcine Growth Hormone and Growth Hormone-Releasing Factor. J. Anim. Sci. 1986, 63, 1389–1399. [Google Scholar] [CrossRef]
- Etherton, T.D.; Wiggins, J.P.; Evock, C.M.; Chung, C.S.; Rebhun, J.F.; Walton, P.E.; Steele, N.C. Stimulation of Pig Growth Performance by Porcine Growth Hormone: Determination of the Dose-Response Relationship. J. Anim. Sci. 1987, 64, 433–443. [Google Scholar] [CrossRef]
- Klindt, J.; Buonomo, F.C.; Yen, J.T.; Pond, W.G.; Mersmann, H.J. Administration of Porcine Somatotropin by Daily Injection: Growth and Endocrine Responses in Genetically Lean and Obese Barrows and Gilts. J. Anim. Sci. 1995, 73, 3294–3303. [Google Scholar] [CrossRef]
- Carakushansky, M.; Whatmore, A.J.; Clayton, P.E.; Shalet, S.M.; Gleeson, H.K.; Price, D.A.; Levine, M.A.; Salvatori, R. A New Missense Mutation in the Growth Hormone-Releasing Hormone Receptor Gene in Familial Isolated GH Deficiency. Eur. J. Endocrinol. 2003, 148, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, E.; Ciappesoni, G.; Iriarte, W.; Da Silva, C.; Macedo, F.; Navajas, E.A.; Brito, G.; San Julian, R.; Gimeno, D.; Postiglioni, A. Novel Genetic Polymorphisms Associated with Carcass Traits in Grazing Texel Sheep. Meat Sci. 2018, 145, 202–208. [Google Scholar] [CrossRef]
- Bakshi, V.P.; Kalin, N.H. Corticotropin-Releasing Hormone and Animal Models of Anxiety: Gene-Environment Interactions. Biol. Psychiatry 2000, 48, 1175–1198. [Google Scholar] [CrossRef]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.L.; Trombitas, K.; Granzier, H.; Gregorio, C.C. Identification of Muscle Specific Ring Finger Proteins as Potential Regulators of the Titin Kinase Domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Borden, K.L. Ring Fingers and B-Boxes: Zinc-Binding Protein-Protein Interaction Domains. Biochem. Cell Biol. 1998, 76, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Freemont, P.S. Ubiquitination: Ring for Destruction? Curr. Biol. 2000, 10, R84–R87. [Google Scholar] [CrossRef] [Green Version]
- Pizon, V.; Iakovenko, A.; Van Der Ven, P.F.; Kelly, R.; Fatu, C.; Furst, D.O.; Karsenti, E.; Gautel, M. Transient Association of Titin and Myosin with Microtubules in Nascent Myofibrils Directed by the MURF2 Ring-Finger Protein. J. Cell Sci. 2002, 115, 4469–4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElhinny, A.S.; Perry, C.N.; Witt, C.C.; Labeit, S.; Gregorio, C.C. Muscle-Specific Ring Finger-2 (MURF-2) Is Important for Microtubule, Intermediate Filament and Sarcomeric M-Line Maintenance in Striated Muscle Development. J. Cell Sci. 2004, 117, 3175–3188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.X.; Li, K.; Liu, B.; Zhu, Z.M.; Xu, X.W.; Zhao, S.H.; Yerle, M.; Fan, B. Chromosomal Localization, Spatio-Temporal Distribution and Polymorphism of the Porcine Tripartite Motif-Containing 55 (TRIM55) Gene. Cytogenet. Genome Res. 2006, 114, 93B. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.; Yakubchyk, Y.; Chabot, J.; Obagi, C.; Daher, E.; Maekawa, K.; Gee, S.H. The Phosphoinositol 3,4-Bisphosphate-Binding Protein TAPP1 Interacts with Syntrophins and Regulates Actin Cytoskeletal Organization. J. Biol. Chem. 2004, 279, 53717–53724. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Song, K.D.; Kim, H.J.; Park, W.; Kim, J.; Lee, T.; Shin, D.H.; Kwak, W.; Kwon, Y.J.; Sung, S.; et al. Exploring the Genetic Signature of Body Size in Yucatan Miniature Pig. PLoS ONE 2015, 10, e0121732. [Google Scholar] [CrossRef] [Green Version]
- Zha, L.; He, L.; Liang, Y.; Qin, H.; Yu, B.; Chang, L.; Xue, L. TNF-α Contributes to Postmenopausal Osteoporosis by Synergistically Promoting RANKL-Induced Osteoclast Formation. Biomed. Pharmacother. 2018, 102, 369–374. [Google Scholar] [CrossRef]
- Zheng, D.; Cui, C.; Yu, M.; Li, X.; Wang, L.; Chen, X.; Lin, Y. Coenzyme Q10 Promotes Osteoblast Proliferation and Differentiation and Protects against Ovariectomy-Induced Osteoporosis. Mol. Med. Rep. 2018, 17, 400–407. [Google Scholar] [CrossRef]
- Hu, Y.; Tan, L.J.; Chen, X.D.; Greenbaum, J.; Deng, H.W. Identification of Novel Variants Associated with Osteoporosis, Type 2 Diabetes and Potentially Pleiotropic Loci Using Pleiotropic cFDR Method. Bone 2018, 117, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Mitin, N.; Rossman, K.L.; Der, C.J. Signaling Interplay in Ras Superfamily Function. Curr. Biol. 2005, 15, R563–R574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ory, S.; Morrison, D.K. Signal Transduction: Implications for Ras-Dependent ERK Signaling. Curr. Biol. 2004, 14, R277–R278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liu, H. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Aouadi, M.; Laurent, K.; Prot, M.; Marchand-Brustel, Y.L.; Binétruy, B.; Bost, F. Inhibition of p38MAPK Increases Adipogenesis from Embryonic to Adult Stages. Diabetes 2006, 55, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wu, W.; Huang, X.; Xu, K.; Zheng, C.; Zhang, J. Comparative Analysis of Gene Expression Profiles in Differentiated Subcutaneous Adipocytes between Jiaxing Black and Large White Pigs. BMC Genom. 2021, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Han, K.; Wu, Y.; Bai, H.; Ke, Q.; Pu, F.; Wang, Y.; Xu, P. Genome-Wide Association Study of Growth and Body-Shape-Related Traits in Large Yellow Croaker (Larimichthys Crocea) Using ddRAD Sequencing. Mar. Biotechnol. 2019, 21, 655–670. [Google Scholar] [CrossRef]
- Lefebvre, V.; Smits, P. Transcriptional Control of Chondrocyte Fate and Differentiation. Birth Defects Res. C Embryo Today 2005, 75, 200–212. [Google Scholar] [CrossRef]
- Kim, S.W.; Jung, J.H.; Do, K.T.; Kim, K.S.; Do, C.H.; Park, J.K.; Joo, Y.K.; Kim, T.S.; Choi, B.H.; Kim, T.H. Investigation of Single Nucleotide Polymorphisms in Porcine Candidate Gene for Growth and Meat Quality Traits in the Berkshire Breed. J. Life Sci. 2007, 17, 1622–1626. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, S.S.; Park, W.C.; Ghosh, M.; Kim, J.N.; Sharma, N.; Shin, K.Y.; Cho, I.C.; Ryu, Y.C.; Oh, S.J.; Kim, S.H.; et al. Comparative Transcriptomic Analysis to Identify Differentially Expressed Genes in Fat Tissue of Adult Berkshire and Jeju Native Pig Using RNA-Seq. Mol. Biol. Rep. 2014, 41, 6305–6315. [Google Scholar] [CrossRef]
- Eppig, J.T.; Group, M.G.D.; Bult, C.J.; Group, M.G.D.; Kadin, J.A.; Group, M.G.D.; Richardson, J.E.; Group, M.G.D.; Blake, J.A.; Group, M.G.D. The Mouse Genome Database (MGD): From Genes to Mice—A Community Resource for Mouse Biology. Nucleic Acids Res. 2005, 33, D471–D475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Lyu, Y.; Zhang, D.; Reddi, K.K.; Sun, F.; Yi, J.; Liu, C.; Li, H.; Yao, H.; Dai, J.; et al. Genomic Characteristics and Selection Signatures in Indigenous Chongming White Goat (Capra Hircus). Front. Genet. 2020, 11, 901. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Lango Allen, H.; Lindgren, C.M.; Luan, J.; Magi, R.; et al. Association Analyses of 249,796 Individuals Reveal 18 New Loci Associated with Body Mass Index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Lee, H.I.; Park, T.; Kim, K.; Lee, J.E.; Cho, N.H.; Shin, C.; Cho, Y.S.; Lee, J.Y.; Han, B.G.; et al. Identification of 15 Loci Influencing Height in A Korean Population. J. Hum. Genet. 2010, 55, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, A.; Forzati, F.; Esposito, F.; Arra, C.; Palma, G.; Barbieri, A.; Palmieri, D.; Fedele, M.; Pierantoni, G.M.; De Martino, I. Hmga1/Hmga2 Double Knock-Out Mice Display A “Superpygmy” Phenotype. Biol. Open 2014, 3, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iiritano, S.; Chiefari, E.; Ventura, V.; Arcidiacono, B.; Possidente, K.; Nocera, A.; Nevolo, M.T.; Fedele, M.; Greco, A.; Greco, M.; et al. The HMGA1-IGF-I/IGFBP System: A Novel Pathway for Modulating Glucose Uptake. Mol. Endocrinol. 2012, 26, 1578–1589. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Walters, G.B.; Thorleifsson, G.; Stefansson, H.; Halldorsson, B.V.; Zusmanovich, P.; Sulem, P.; Thorlacius, S.; Gylfason, A.; Steinberg, S.; et al. Many Sequence Variants Affecting Diversity of Adult Human Height. Nat. Genet. 2008, 40, 609–615. [Google Scholar] [CrossRef]
- Zhang, L.C.; Li, N.; Liu, X.; Liang, J.; Yan, H.; Zhao, K.B.; Pu, L.; Shi, H.B.; Zhang, Y.B.; Wang, L.G. A Genome-Wide Association Study of Limb Bone Length Using A Large White × Minzhu Intercross Population. Genet. Sel. Evol. 2014, 46, 56. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into Lafora Disease: Malin Is an E3 Ubiquitin Ligase That Ubiquitinates and Promotes the Degradation of Laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [Green Version]
- Lohi, H.; Ianzano, L.; Zhao, X.C.; Chan, E.M.; Turnbull, J.; Scherer, S.W.; Ackerley, C.A.; Minassian, B.A. Novel Glycogen Synthase Kinase 3 and Ubiquitination Pathways in Progressive Myoclonus Epilepsy. Hum. Mol. Genet. 2005, 14, 2727–2736. [Google Scholar] [CrossRef]
- Solaz-Fuster, M.C.; Gimeno-Alcaniz, J.V.; Ros, S.; Fernandez-Sanchez, M.E.; Garcia-Fojeda, B.; Garcia, O.C.; Vilchez, D.; Dominguez, J.; Garcia-Rocha, M.; Sanchez-Piris, M. Regulation of Glycogen Synthesis by the Laforin–Malin Complex Is Modulated by the AMP-Activated Protein Kinase Pathway. Hum. Mol. Genet. 2008, 17, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illa, S.K.; Mukherjee, S.; Nath, S.; Mukherjee, A. Genome-Wide Scanning for Signatures of Selection Revealed the Putative Genomic Regions and Candidate Genes Controlling Milk Composition and Coat Color Traits in Sahiwal Cattle. Front. Genet. 2021, 12, 699422. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wang, Z.; Zhang, Q.; Yue, S.; Yin, B.; Jiang, Y.; Shi, K. Identification of Whole-Genome Significant Single Nucleotide Polymorphisms in Candidate Genes Associated with Body Conformation Traits in Chinese Holstein Cattle. Anim. Genet. 2020, 51, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.W.; Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Tripartite-Motif Proteins and Innate Immune Regulation. Curr. Opin. Immunol. 2011, 23, 46–56. [Google Scholar] [CrossRef]
- Li, W.; Zheng, M.; Zhao, G.; Wang, J.; Liu, J.; Wang, S.; Feng, F.; Liu, D.; Zhu, D.; Li, Q.; et al. Identification of QTL Regions and Candidate Genes for Growth and Feed Efficiency in Broilers. Genet. Sel. Evol. 2021, 53, 13. [Google Scholar] [CrossRef]
- Yang, H.; Sasaki, T.; Minoshima, S.; Shimizu, N. Identification of Three Novel Proteins (SGSM1, 2, 3) Which Modulate Small G Protein (RAP and RAB)-Mediated Signaling Pathway. Genomics 2007, 90, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhao, W.; Fu, Y.; Fang, X.; Ren, S.; Ren, J. Genome-Wide Detection of Genetic Loci and Candidate Genes for Teat Number and Body Conformation Traits at Birth in Chinese Sushan Pigs. Anim. Genet. 2019, 50, 753–756. [Google Scholar] [CrossRef]
- Horsley, V.; Pavlath, G.K. Prostaglandin F2α Stimulates Growth of Skeletal Muscle Cells Via an NFATC2-Dependent Pathway. J. Cell Biol. 2003, 161, 111–118. [Google Scholar] [CrossRef]
- Pavlath, G.K.; Horsley, V. Cell Fusion in Skeletal Muscle--Central Role of NFATC2 in Regulating Muscle Cell Size. Cell Cycle 2003, 2, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Karthikeyan, A.; Kumar, A.; Chaudhary, R.; Wara, A.B.; Singh, A.; Sahoo, N.; Baqir, M.; Mishra, B. Genome-Wide Association Study of Birth Weight and Pre-Weaning Body Weight of Crossbred Pigs. Indian J. Anim. Sci. 2020, 90, 195–200. [Google Scholar] [CrossRef]
- Dusseault, J.; Li, B.; Haider, N.; Goyette, M.A.; Côté, J.F.; Larose, L. Nck2 Deficiency in Mice Results in Increased Adiposity Associated with Adipocyte Hypertrophy and Enhanced Adipogenesis. Diabetes 2016, 65, 2652–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClure, M.C.; Morsci, N.S.; Schnabel, R.D.; Kim, J.W.; Yao, P.; Rolf, M.M.; McKay, S.D.; Gregg, S.J.; Chapple, R.H.; Northcutt, S.L.; et al. A Genome Scan for Quantitative Trait Loci Influencing Carcass, Post-Natal Growth and Reproductive Traits in Commercial Angus Cattle. Anim. Genet. 2010, 41, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; McElhinny, A.S.; Cao, Y.; Gao, P.; Liu, J.; Bronson, R.; Griffin, J.D.; Wu, L. The Notch Coactivator, MAML1, Functions as a Novel Coactivator for MEF2C-Mediated Transcription and Is Required for Normal Myogenesis. Genes Dev. 2006, 20, 675–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeyemo, A.; Luke, A.; Cooper, R.; Wu, X.D.; Tayo, B.; Zhu, X.F.; Rotimi, C.; Bouzekri, N.; Ward, R. A Genome-Wide Scan for Body Mass Index among Nigerian Families. Obesity 2003, 11, 266–273. [Google Scholar] [CrossRef]
- Saar, K.; Geller, F.; Ruschendorf, F.; Reis, A.; Friedel, S.; Schauble, N.; Nurnberg, P.; Siegfried, W.; Goldschmidt, H.P.; Schafer, H. Genome Scan for Childhood and Adolescent Obesity in German Families. Pediatrics 2003, 111, 321–327. [Google Scholar] [CrossRef]
- Gong, S.; Xu, C.; Wang, L.; Liu, Y.; Owusu, D.; Bailey, B.A.; Li, Y.; Wang, K. Genetic Association Analysis of Polymorphisms in PSD3 Gene with Obesity, Type 2 Diabetes, and HDL Cholesterol. Diabetes Res. Clin. Pract. 2017, 126, 105–114. [Google Scholar] [CrossRef]
- Luo, J.; Shen, L.Y.; Gan, M.; Jiang, A.A.; Chen, L.; Ma, J.D.; Jin, L.; Liu, Y.H.; Tang, G.Q.; Jiang, Y.Z.; et al. Profiling of Skeletal Muscle Tissue for Long Non-Coding RNAs Related to Muscle Metabolism in the QingYu Pig at the Growth Inflection Point. Anim. Biosci. 2021, 34, 1309. [Google Scholar] [CrossRef]
- Swatland, H.J.; Cassens, R.G. Prenatal Development, Histochemistry and Innervation of Porcine Muscle. J. Anim. Sci. 1973, 36, 343–354. [Google Scholar] [CrossRef]
- Zhao, Y.Q.; Li, J.; Liu, H.j.; Xi, Y.; Xue, M.; Liu, W.H.; Zhuang, Z.H.; Lei, M.G. Dynamic Transcriptome Profiles of Skeletal Muscle Tissue Across 11 Developmental Stages for Both Tongcheng and Yorkshire Pigs. BMC Genom. 2015, 16, 377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Y.; Wang, H.Y.; Tu, W.L.; Raza, S.H.A.; Cao, J.G.; Huang, J.; Wu, H.L.; Fan, C.; Wang, S.C.; Zhao, Y. Comparative Transcriptome Analysis Provides Insight into Spatio-Temporal Expression Characteristics and Genetic Regulatory Network in Postnatal Developing Subcutaneous and Visceral Fat of Bama Pig. Front. Genet. 2022, 13, 844833. [Google Scholar] [CrossRef]
- Etherton, T.D.; Walton, P.E. Hormonal and Metabolic Regulation of Lipid Metabolism in Domestic Livestock. J. Anim. Sci. 1986, 63, 76–88. [Google Scholar] [CrossRef]
- Lai, K.M.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional Activation of Akt in Adult Skeletal Muscle Induces Rapid Hypertrophy. Mol. Cell Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait * | LBT | ADG30–60 | ADG60–100 | ADG30–100 | BL | BH | BAH | CC | CD |
|---|---|---|---|---|---|---|---|---|---|
| ADG30–60 | 0.29 *** | ||||||||
| ADG60–100 | 0.19 * | −0.03 | |||||||
| ADG30–100 | 0.29 *** | 0.58 *** | 0.76 *** | ||||||
| BL | 0.01 | 0.18 * | 0.25 *** | 0.34 *** | |||||
| BH | −0.07 | 0.04 | 0.17 * | 0.20 ** | 0.41 *** | ||||
| BAH | 0.06 | 0.18 * | 0.23 ** | 0.36 *** | 0.54 *** | 0.70 *** | |||
| CC | 0.10 | 0.04 | 0.28 *** | 0.25 *** | 0.26 *** | 0.21 ** | 0.17 ** | ||
| CD | −0.12 * | −0.15 * | 0.10 | −0.02 | −0.06 | 0.33 *** | 0.20 ** | 0.12 * | |
| RC | 0.01 | 0.05 | 0.16 * | 0.24 *** | 0.46 *** | 0.21 ** | 0.37 *** | 0.34 *** | −0.12 * |
| Trait Pair * | Genetic Correlation | Standard Error |
|---|---|---|
| ADG30–60/ADG60–100 | −0.27 | 0.13 |
| ADG30–60/ADG30–100 | 0.87 | 0.10 |
| ADG60–100/ADG30–100 | 0.93 | 0.05 |
| Trait a | SNP b | SSC c | Position (bp) d | Genotype e | N f | Value g | Additive Effect | Dominance Effect |
|---|---|---|---|---|---|---|---|---|
| ADG30–60 (g) | rs325760894 | 4 | 68428526 | GG | 34 | 536.39 ± 18.59 A | 75.63 | −4.36 |
| GT | 101 | 456.43 ± 10.79 B | ||||||
| TT | 57 | 385.13 ± 14.36 C | ||||||
| NN | 6 | |||||||
| rs81382100 | 4 | 68463657 | CC | 67 | 384.42 ± 13.24 C | 67.43 | 14.90 | |
| CT | 85 | 466.75 ± 11.76 B | ||||||
| TT | 42 | 519.28 ± 16.72 A | ||||||
| NN | 4 | |||||||
| rs320502793 | 4 | 68493463 | AA | 58 | 382.11 ± 14.14 C | 69.53 | 0.70 | |
| AG | 84 | 452.34 ± 11.75 B | ||||||
| GG | 52 | 521.17 ± 14.93 A | ||||||
| NN | 4 | |||||||
| ADG60–100 (g) | 14 | 131969638 | CC | 146 | 545.88 ± 9.88 cB | 88.24 | −21.13 | |
| CT | 19 | 637.99 ± 27.40 bA | ||||||
| TT | 4 | 772.36 ± 59.71 aA | ||||||
| NN | 29 | |||||||
| BAH (cm) | 14 | 42805887 | CC | 138 | 71.44 ± 0.33 A | 2.88 | 0.67 | |
| CT | 35 | 69.23 ± 0.65 B | ||||||
| TT | 13 | 65.69 ± 1.07 C | ||||||
| NN | 37 | |||||||
| 14 | 42805901 | AA | 153 | 71.16 ± 0.32 aA | 3.37 | 1.38 | ||
| AT | 24 | 69.17 ± 0.81 bA | ||||||
| TT | 7 | 64.43 ± 1.50 cB | ||||||
| NN | 39 | |||||||
| CD (cm) | rs705385434 | 2 | 78815567 | GG | 180 | 36.06 ± 0.18 A | 3.47 | −1.24 |
| GT | 21 | 38.29 ± 0.53 B | ||||||
| TT | 3 | 43.00 ± 1.40 C | ||||||
| NN | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wang, X.; Li, M.; Sun, H.; Chen, Q.; Yan, D.; Dong, X.; Pan, Y.; Lu, S. Genome-Wide Association Study of Growth Traits in a Four-Way Crossbred Pig Population. Genes 2022, 13, 1990. https://doi.org/10.3390/genes13111990
Wang H, Wang X, Li M, Sun H, Chen Q, Yan D, Dong X, Pan Y, Lu S. Genome-Wide Association Study of Growth Traits in a Four-Way Crossbred Pig Population. Genes. 2022; 13(11):1990. https://doi.org/10.3390/genes13111990
Chicago/Turabian StyleWang, Huiyu, Xiaoyi Wang, Mingli Li, Hao Sun, Qiang Chen, Dawei Yan, Xinxing Dong, Yuchun Pan, and Shaoxiong Lu. 2022. "Genome-Wide Association Study of Growth Traits in a Four-Way Crossbred Pig Population" Genes 13, no. 11: 1990. https://doi.org/10.3390/genes13111990