
Metabolomic and Transcriptomic Analyses Reveal the Characteristics of Tea Flavonoids and Caffeine Accumulation and Regulation between Chinese Varieties (Camellia sinensis var. sinensis) and Assam Varieties (C. sinensis var. assamica)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Metabolite Extraction and MS Detection
2.3. MS Data and Statistical Analyses
2.4. Library Preparation for RNA Sequencing
2.5. Function Annotation and Expression Analysis
2.6. Measurements of Other Main Metabolites
3. Results
3.1. Comparison of Morphological Phenotype and Metabolomic Analysis
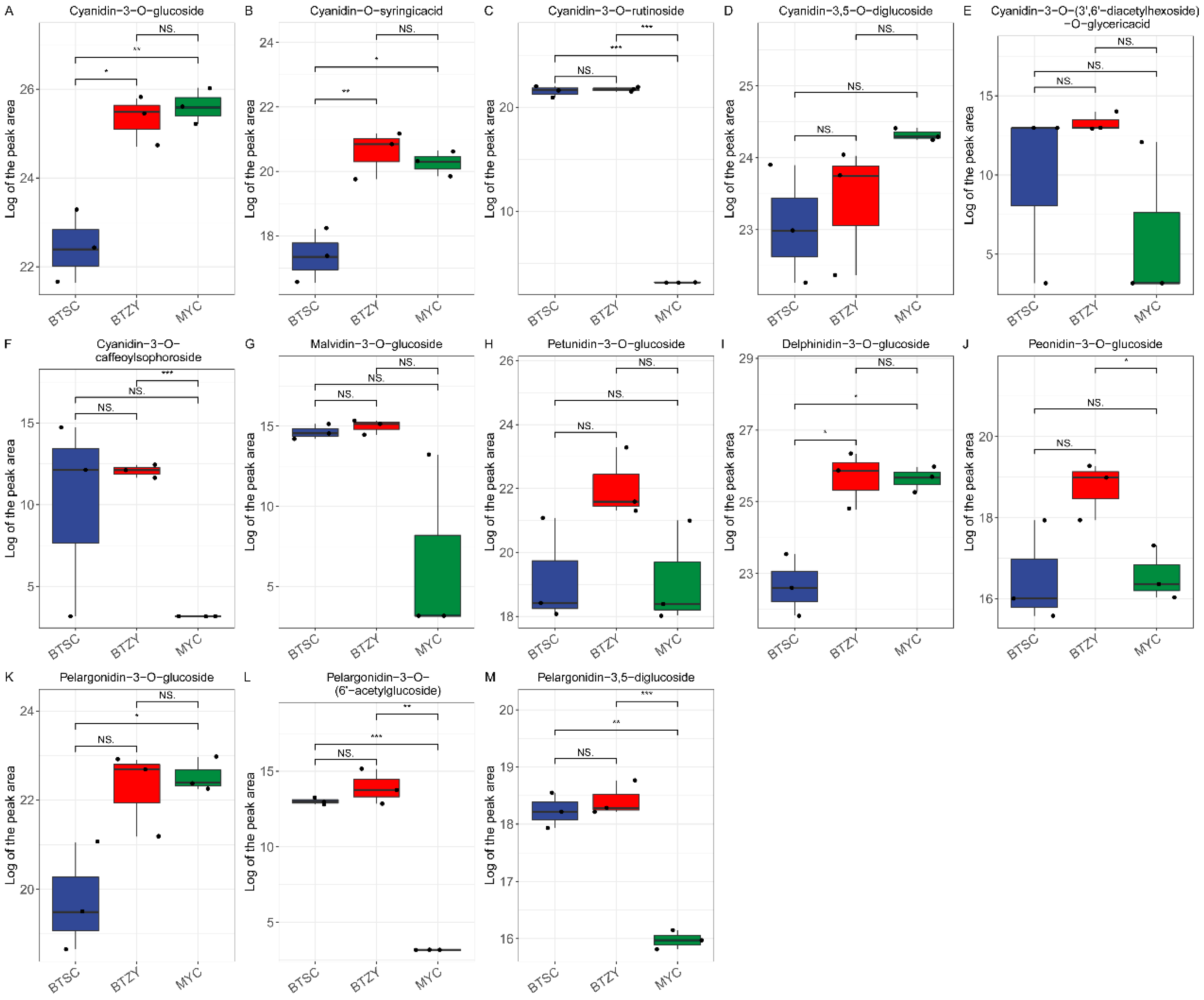
3.2. Differential Accumulated Metabolites Analyses
3.3. Measurement of Bioactive Components
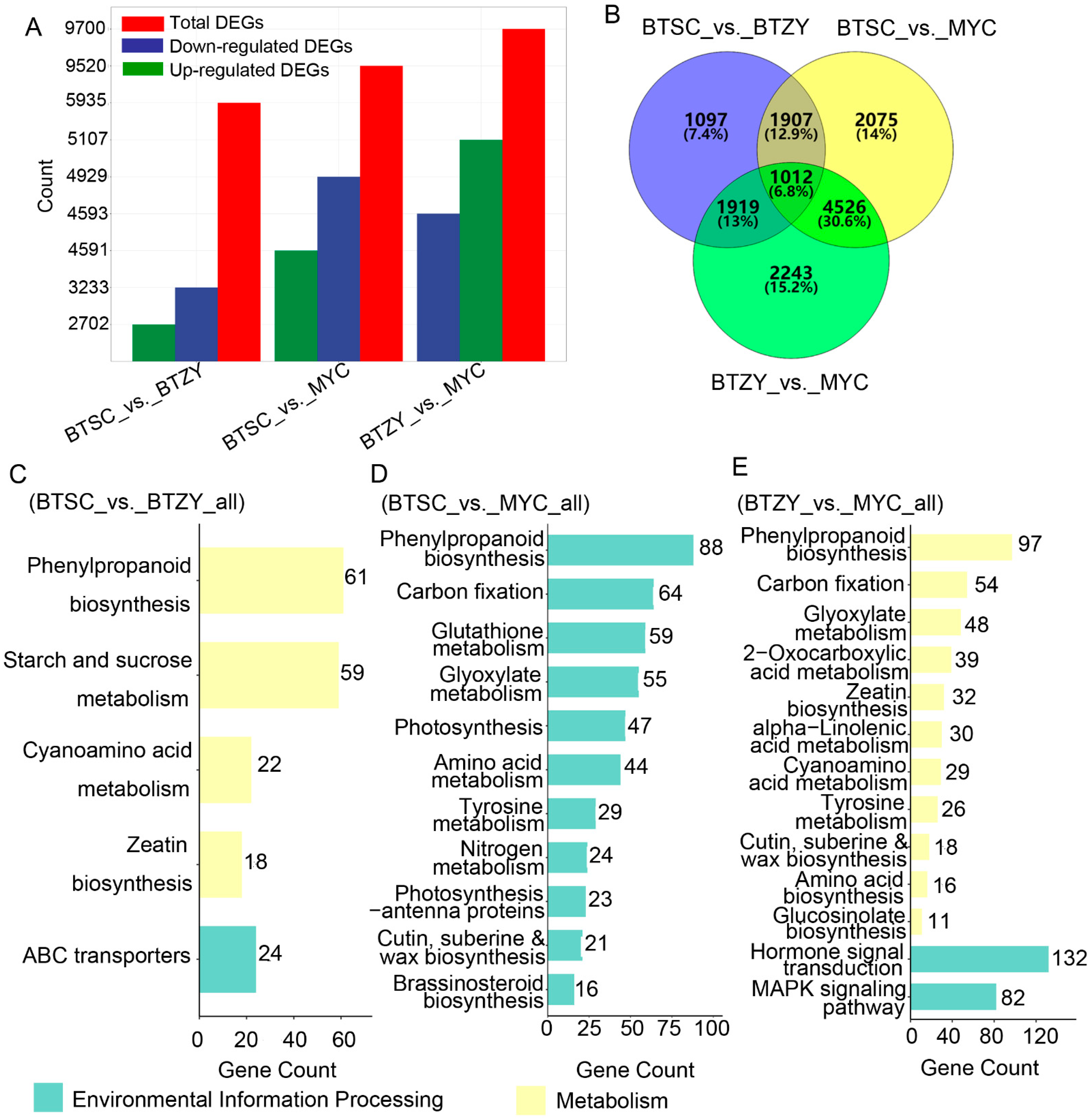
3.4. Identification of Differentially Expressed Genes
3.5. Functional Annotation of Differentially Expressed Genes
3.6. Expression of the Genes on Anthocyanin and Catechin Biosynthesis
3.7. Regulation of Caffeine Biosynthesis in ‘MYC’ Group
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mondal, T.K.; Bhattacharya, A.; Laxmikumaran, M.; Ahuja, P.S. Recent advances of tea (Camellia sinensis) biotechnology. Plant Cell Tissue Organ Cult. 2004, 76, 195–254. [Google Scholar] [CrossRef]
- Fan, Y.; Zhou, X.; Huang, G. Preparation, structure, and properties of tea polysaccharide. Chem. Biol. Drug Des. 2022, 99, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cheng, S.; Dai, J.; Wang, L.; Xu, Y.; Peng, X.; Xie, X.; Peng, C. Molecular mechanisms and applications of tea polyphenols: A narrative review. J. Food Biochem. 2021, 45, e13910. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Y.; Gan, R.Y.; Zhu, F. Chemical constituents and biological properties of Puerh tea. Food Res. Int. 2022, 154, 110899. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Li, J.; Huang, J.A.; Liu, Z.H.; Xiong, L.G. New advances in genetic engineering for l-theanine biosynthesis. Trends Food Sci. Technol. 2021, 114, 540–551. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar]
- Tomohiro, A.; Yoshitaka, H.; Toshiyuki, K. Chemical synthesis of tea polyphenols and related compounds. Curr. Pharm. Des. 2013, 19, 6207–6217. [Google Scholar]
- Pomier, K.M.; Ahmed, R.; Melacini, G. Catechins as tools to understand the molecular basis of neurodegeneration. Molecules 2020, 25, 3571. [Google Scholar] [CrossRef]
- Minnelli, C.; Cianfruglia, L.; Laudadio, E.; Mobbili, G.; Galeazzi, R.; Armeni, T. Effect of epigallocatechin-3-gallate on EGFR signaling and migration in non-small cell lung cancer. Int. J. Mol. Sci. 2021, 22, 11833. [Google Scholar] [CrossRef]
- He, H.F. Research progress on theaflavins: Efficacy, formation, and preparation. Food Nutr. Res. 2017, 61, 1344521. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Zhang, C.; Shao, C.; Liu, B.; Liu, E.; Yuan, D.; Zhou, Y.; Shen, C. Research progress on the response of tea catechins to drought stress. J. Sci. Food Agric. 2021, 101, 5305–5313. [Google Scholar] [CrossRef] [PubMed]
- Paiva, L.; Rego, C.; Lima, E.; Marcone, M.; Baptista, J. Comparative Analysis of the polyphenols, caffeine, and antioxidant activities of green tea, white tea, and flowers from azorean Camellia sinensis varieties affected by different harvested and processing conditions. Antioxidants 2021, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Unno, K.; Noda, S.; Kawasaki, Y.; Yamada, H.; Morita, A.; Iguchi, K.; Nakamura, Y. Reduced stress and improved sleep quality caused by green tea are associated with a reduced caffeine content. Nutrients 2017, 9, 777. [Google Scholar] [CrossRef]
- Jee, H.J.; Lee, S.G.; Bormate, K.J.; Jung, Y.S. Effect of caffeine consumption on the risk for neurological and psychiatric disorders: Sex differences in human. Nutrients 2020, 12, 3080. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.H.; Zhang, H.B.; Sheng, J.; Li, K.; Zhang, Q.J.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, D.; Zhang, H.; Lai, B.; Liu, L.; Pan, X.; Ma, Z.; Wang, Y.; Xie, J.; Shi, S.; Wei, Y. Integrative analysis of the coloring mechanism of red longan pericarp through metabolome and transcriptome analyses. J. Agric. Food Chem. 2021, 69, 9. [Google Scholar] [CrossRef]
- Sun, B.; Zhu, Z.; Cao, P.; Chen, H.; Chen, C.; Zhou, X.; Mao, Y.; Lei, J.; Jiang, Y.; Meng, W.; et al. Purple foliage coloration in tea (Camellia sinensis L.) arises from activation of the R2R3-MYB transcription factor CsAN1. Sci. Rep. 2016, 6, 32534. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Fu, J.; Xu, Y.; Shen, Y.; Zhang, Y.; Ye, Z.; Tong, W.; Zeng, X.; Yang, J.; Tang, D.; et al. CsMYB1 integrates the regulation of trichome development and catechins biosynthesis in tea plant domestication. New Phytol. 2022, 234, 902–917. [Google Scholar] [CrossRef]
- Mei, Y.; Xie, H.; Liu, S.; Zhu, J.; Zhao, S.; Wei, C. Metabolites and transcriptional profiling analysis reveal the molecular mechanisms of the anthocyanin metabolism in the “Zijuan” tea plant (Camellia sinensis var. assamica). J. Agric. Food Chem. 2021, 69, 414–427. [Google Scholar] [CrossRef]
- Maritim, T.K.; Masand, M.; Seth, R.; Sharma, R.K. Transcriptional analysis reveals key insights into seasonal induced anthocyanin degradation and leaf color transition in purple tea (Camellia sinensis (L.) O. Kuntze). Sci. Rep. 2021, 11, 1244. [Google Scholar] [CrossRef]
- Jin, J.Q.; Yao, M.Z.; Ma, C.L.; Ma, J.Q.; Chen, L. Natural allelic variations of TCS1 play a crucial role in caffeine biosynthesis of tea plant and its related species. Plant Physiol. Biochem. 2016, 100, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ye, Z.; Fu, J.; Xu, Y.; Shen, Y.; Zhang, Y.; Tang, D.; Li, P.; Zuo, H.; Tong, W.; et al. CsMYB184 regulates caffeine biosynthesis in tea plants. Plant Biotechnol. J. 2022, 20, 1012–1014. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Wu, J.; Shahid, M.Q.; He, Y.; Lin, S.; Liu, Z.; Yang, X. Identification of key taste components in loquat using widely targeted metabolomics. Food Chem. 2020, 323, 126822. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, L.; Huang, X.; Wang, X.; Yang, R.; Mao, J.; Wang, X.; Wang, X.; Zhang, Q.; Li, P. Identification of nutritional components in black sesame determined by widely targeted metabolomics and traditional chinese medicines. Molecules 2018, 23, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Huang, Y.; Wu, W.; Zhu, C.; Zhang, R.; Chen, J.; Zeng, J. Metabolomics analysis of the peels of different colored citrus fruits (Citrus reticulata cv. ‘Shatangju’) during the maturation period based on UHPL-QQQ-MS. Molecules 2020, 25, 396. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Chen, L.B.; Lu, M.; Zhang, J.; Han, J.; Deng, W.W.; Zhang, Z.Z. Caffeine content and related gene expression: Novel insight into caffeine metabolism in Camellia plants containing low, normal, and high caffeine concentrations. J. Agric. Food Chem. 2019, 67, 3400–3411. [Google Scholar] [CrossRef]
- Zheng, X.L.; Tian, S.P. Effect of oxalic acid on control of postharvest browning of litchi fruit. Food Chem. 2006, 96, 519–523. [Google Scholar] [CrossRef]
- Chen, X.; Wang, P.; Zheng, Y.; Gu, M.; Lin, X.; Wang, S.; Jin, S.; Ye, N. Comparison of metabolome and transcriptome of flavonoid biosynthesis pathway in a purple-leaf tea germplasm Jinmingzao and a green-leaf tea germplasm Huangdan reveals their relationship with genetic mechanisms of color formation. Int. J. Mol. Sci. 2020, 21, 4167. [Google Scholar] [CrossRef]
- Li, J.; Hossain, M.S.; Ma, H.; Yang, Q.; Gong, X.; Yang, P.; Feng, B. Comparative metabolomics reveals differences in flavonoid metabolites among different coloured buckwheat flowers. J. Food Compos. Anal. 2020, 85, 103335. [Google Scholar] [CrossRef]
- Cai, J.; Lv, L.; Zeng, X.; Zhang, F.; Chen, Y.; Tian, W.; Li, J.; Li, X.; Li, Y. Integrative analysis of metabolomics and transcriptomics reveals molecular mechanisms of anthocyanin metabolism in the Zikui tea plant (Camellia sinensis cv. Zikui). Int. J. Mol. Sci. 2022, 23, 4780. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Wang, L.; Zhang, Y.; Ruan, L.; Li, H.; Wu, L.; Xu, L.; Zhang, C.; Zhou, X.; Cheng, H.; et al. A coupled role for CsMYB75 and CsGSTF1 in anthocyanin hyperaccumulation in purple tea. Plant J. 2019, 97, 825–840. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, C.; Lu, M.; Gan, N.; Chen, Z.; Deng, W.W.; Zhang, Z.Z. Metabolic flux enhancement and transcriptomic analysis displayed the changes of catechins following long-term pruning in tea trees (Camellia sinensis). J. Agric. Food Chem. 2018, 66, 8566–8573. [Google Scholar] [CrossRef] [PubMed]
- Mugford, S.T.; Osbourn, A. Evolution of serine carboxypeptidase-like acyltransferases in the monocots. Plant Signal. Behav. 2010, 5, 193–195. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gao, L.; Liu, L.; Yang, Q.; Lu, Z.; Nie, Z.; Wang, Y.; Xia, T. Purification and characterization of a novel galloyltransferase involved in catechin galloylation in the tea plant (Camellia sinensis). J. Biol. Chem. 2012, 287, 44406–44417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.; Wang, L.Y.; Zhou, J.; He, W.; Zeng, J.M.; Jiang, Y.W.; Cheng, H. Comparison of catechins and purine alkaloids in albino and normal green tea cultivars (Camellia sinensis L.) by HPLC. Food Chem. 2012, 130, 720–724. [Google Scholar] [CrossRef]
- Kato, M.; Kanehara, T.; Shimizu, H.; Suzuki, T.; Gillies, F.M.; Crozier, A.; Ashihara, H. Caffeine biosynthesis in young leaves of Camellia sinensis: In vitro studies on N-methyltransferase activity involved in the conversion of xanthosine to caffeine. Physiol. Plant. 1996, 98, 629–636. [Google Scholar] [CrossRef]
- Kato, M.; Mizuno, K.; Crozier, A.; Fujimura, T.; Ashihara, H. Caffeine synthase gene from tea leaves. Nature 2000, 406, 956–957. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Hu, B.; Qin, Y.; Hu, G.; Zhao, J. Advance of the negative regulation of anthocyanin biosynthesis by MYB transcription factors. Plant Physiol. Biochem. 2019, 136, 178–187. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Zhang, M.; Liu, J.; Cai, J. Metabolomic and Transcriptomic Analyses Reveal the Characteristics of Tea Flavonoids and Caffeine Accumulation and Regulation between Chinese Varieties (Camellia sinensis var. sinensis) and Assam Varieties (C. sinensis var. assamica). Genes 2022, 13, 1994. https://doi.org/10.3390/genes13111994
Tang H, Zhang M, Liu J, Cai J. Metabolomic and Transcriptomic Analyses Reveal the Characteristics of Tea Flavonoids and Caffeine Accumulation and Regulation between Chinese Varieties (Camellia sinensis var. sinensis) and Assam Varieties (C. sinensis var. assamica). Genes. 2022; 13(11):1994. https://doi.org/10.3390/genes13111994
Chicago/Turabian StyleTang, Hao, Man Zhang, Jiayu Liu, and Jiao Cai. 2022. "Metabolomic and Transcriptomic Analyses Reveal the Characteristics of Tea Flavonoids and Caffeine Accumulation and Regulation between Chinese Varieties (Camellia sinensis var. sinensis) and Assam Varieties (C. sinensis var. assamica)" Genes 13, no. 11: 1994. https://doi.org/10.3390/genes13111994