1. Introduction
Long-non-coding RNAs (lncRNAs) are long chains composed of nucleotides, with a wide range of actions and complex mechanisms. They get involved in many critical regulatory processes [
1,
2,
3,
4] and have attracted the attention of many life scientists and biologists in recent years. Studies have found that mutations and disorders of lncRNAs are bound up with the occurrence of human diseases [
5,
6], including AIDS [
7], diabetes [
8], Alzheimer’s disease [
9], and many types of cancer, such as breast cancer [
10], prostate [
11], hepatocellular [
12], and bladder cancer [
13]. Many associations between lncRNAs and diseases and how they interact have also become a good breakthrough for researchers to understand the pathogenesis of diseases from the molecular level.
Although the research on identifying human lncRNA-disease associations (LDAs) progresses rapidly, the precise principles behind it remain largely unclear, such as transcriptional regulation, multi-biological processes, and molecular mechanisms of various diseases [
14]. Predicting the undiscovered LDAs can help people figure out the pivotal factor of lncRNAs in biological processes, thus helping with the diagnosis, treatment, and prognosis of diseases. Using computational models to predict potential LDAs takes far less time and cost than biological experiments. Therefore, it is of great significance to study computational models to reveal new LDAs for further experimental verification. Scientists have done a lot to the research of lncRNA-disease relationship, and many excellent predictive models have appeared [
15,
16,
17]. Existing models for predicting LDAs mainly fall into two categories: machine learning-based methods and biological network-based methods [
18]. Machine learning-based methods play an important role in predicting LDAs. Classifiers can be trained based on the characteristics of known disease-associated lncRNAs and those of unknown disease-associated lncRNAs. Candidate lncRNAs can be ranked in line with the differences of biological characteristics. Lan et al. [
19] developed a supervised method: LDAP, which integrated multivariate biological data. In this method, the bagging support vector machine (SVM) was trained to predict LDAs. Multiple training datasets are constructed by bagging method, and each dataset is trained by SVM to generate multiple weak classifiers, which vote on the category of test samples. Chen et al. [
20] proposed a computational method: Laplacian Regularized Least Squares for LDA (LRLSLDA). This method was based on a semi-supervised learning framework to predict new LDAs and achieved reliable performance. However, LRLSLDA still has some limitations. For example, there are many parameters in the method, and it is very difficult to determine the optimal parameters. In addition, for the same LDA pair, two different scores can be obtained from the lncRNA space and the disease space, respectively. How to efficiently combine the two scores has become a current research topic. Gao et al. designed a method: Multi-Label Fusion Collaborative matrix factorization (MLFCMF) [
21] to identify LDAs. First, the inner links between lncRNAs and diseases were improved and the hidden information was discovered by multi-label learning. Second, the fusion method was used to learn the multi-label information. Finally, potential LDAs were inferred by collaborative matrix factorization. Fu et al. [
17] reconstructed the LDA matrix by the optimized low-rank matrices to identify latent LDAs. Lu et al. [
22] proposed a method to recover informative features by principle components analysis and complement the LDA matrix derived from the inductive matrix completion. For the machine learning-based methods, the main challenge is how to select useful biometrics to train the classifier. Therefore, integrating multiple data resources can effectively improve prediction performance. Biswas et al. [
23] designed a novel method for predicting potential LDAs based on matrix factorization. The model integrated known LDAs, experimentally verified gene-disease associations, gene-gene interaction data, and the profiles of lncRNAs and genes. The bi-clustering method was used to identify lncRNA modules and non-negative matrix factorization (NMF) was used to reveal potential LDAs.
In recent years, the outstanding performance of network-based methods in predicting LDAs has aroused the researchers’ interest. Many excellent algorithms have emerged based on the hypothesis that functionally similar lncRNAs may be related to diseases with similar phenotypes. For example, Sun et al. [
24] proposed a computing method, namely RWRlncD. In this study, after the establishment of the LDA network, the disease similarity network (DSN) and the lncRNA similarity network (LSN), RWRlncD predicted the potential LDAs by randomly walking on the LSN. It is worth noting that RWRlncD is robust to different parameters. As more LDAs and more accurate measures of the lncRNA functional similarity become available, the prediction ability of RWRlncD will be improved. Zhou et al. [
25] also designed a novel model to identify potential LDAs. This model integrated three networks (i.e., the miRNA-associated lncRNA-lncRNA crosstalk network, the DSN and the known LDA network) into one network and conducted random walks on it. However, the method is only applicable to lncRNAs with known lncRNA–miRNA interactions. In addition, the incomplete coverage of the lncRNAs crosstalk network and the LDA network may reduce the prediction performance of the model. Xie et al. [
26] developed a method to infer new LDAs. First, the features of lncRNAs and diseases were mapped to the features of local-constraint by location-constrained linear coding, and then the initial correlation matrix and the acquired features of lncRNAs and diseases were mixed up by the label propagation strategy. Xie et al. [
18] also used the weighted K-nearest known neighbors algorithm (WKNKN) method to solve the problem with rare known LDAs and applied the linear neighbor similarity (LNS) to reconstruct the DSN and LSN. In 2020, Ref. [
27] designed a method to reveal potential LDAs. The method combined the heat spread algorithm and probability diffusion algorithm to reallocate resources, and used unbalanced bi-random walks to infer new LDAs.
However, these methods have some drawbacks. For example, most methods only introduce Gaussian Interaction Profile (GIP) kernel similarity, which makes the prior information used for prediction too simple and single. In response to this question, we propose a new method called MSF-UBRW to infer potential LDAs based on multiple similarities fusion and unbalanced bi-random walk. First, the lncRNA functional similarity matrix is obtained from known LDA matrix. Second, the GIP kernel similarity of lncRNAs is calculated derived from known LDAs, and the logistic function is used to adjust the similarity of the lncRNA network. The same is true for the disease network. Third, linear fusion is performed for the above two similarities of lncRNAs and diseases, respectively. Then, the initial association probability matrix is calculated by WKNKN. Next, the pairwise linear neighborhood similarities of lncRNAs and diseases are calculated. Finally, LDAs are inferred by bi-randomly walking with different steps on the lncRNA network and the disease network. The main highlights of the MSF-UBRW method are as follows:
(1) Linear fusion was performed for lncRNA functional similarity and GIP kernel similarity of lncRNAs, as well as for disease semantic similarity and GIP kernel similarity of diseases. In addition to that, logistic functions are constructed from known LDAs to improve the topology structure of networks.
(2) So far, very few LDAs have been identified, which results in a sparse LDA matrix. WKNKN is used to preprocess the known LDA matrix to solve the sparse problem and obtain the association probability matrix.
(3) The linear neighbor similarity is applied to reconstruct the DSN and LSN.
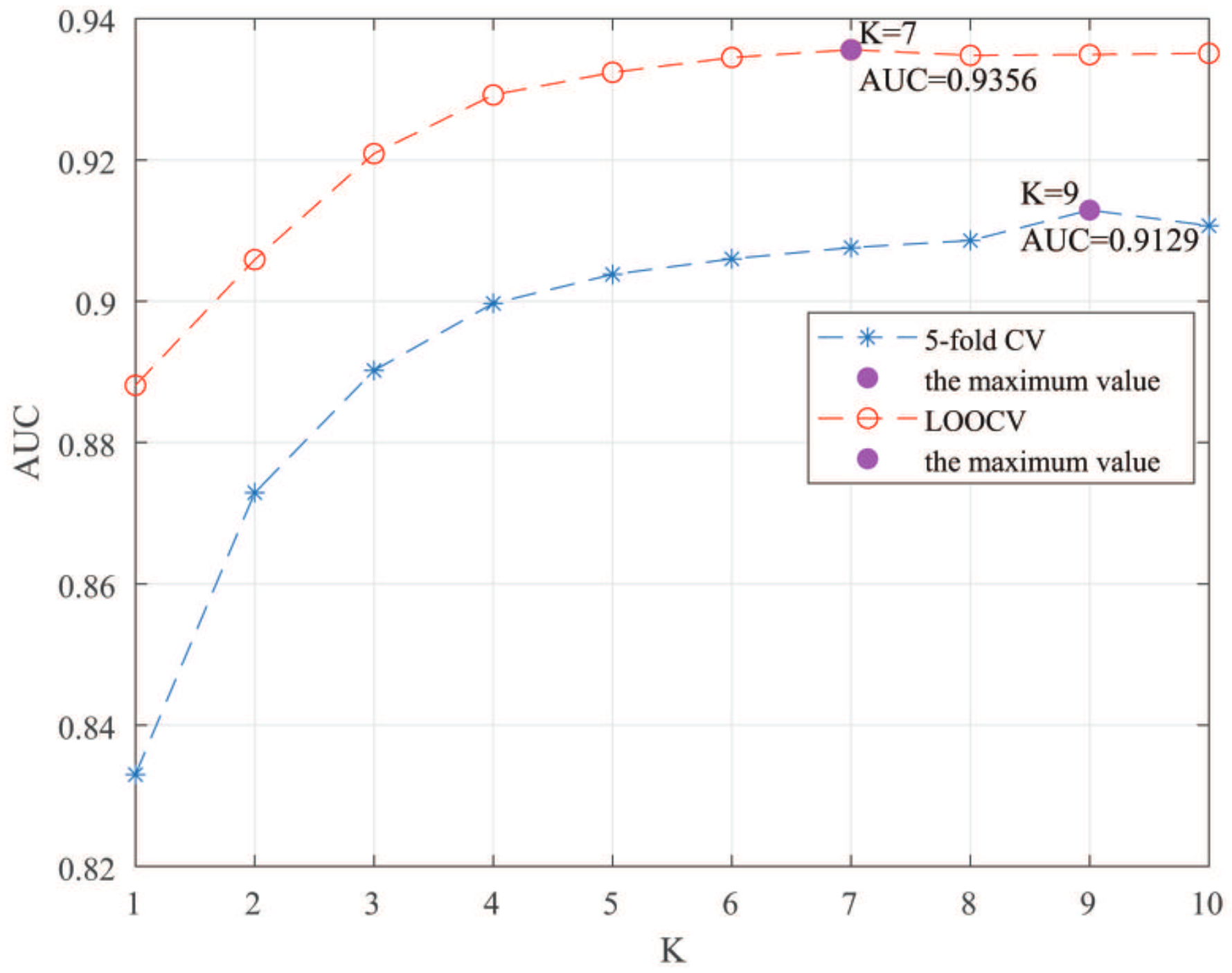
The MSF-UBRW method achieves the reliable AUC values with and 0.9183 based on leave-one-out cross validation (LOOCV) and 5-fold cross validation (5-fold CV), respectively. In addition, case studies of three common diseases (prostate cancer, esophageal squamous cell carcinoma (ESCC), and small cell lung cancer (NSCLC)) further prove the prediction ability of the MSF-UBRW method. Experimental results demonstrate that MSF-UBRW is an effective and reliable method for identifying potential LDAs.
4. Conclusions
More and more studies have found that changes in lncRNA expression patterns are associated with specific diseases. Building computational models to predict LDAs is not only a meaningful complement to experimental methods, but also helps researchers to gain insight into the pathogenesis of diseases. In this study, based on GIP and LNS, MSF-UBRW performs unbalanced bi-random walks in the LSN and DSN based on multiple similarities fusion to find new LDAs. Compared with LDA-LNSUBRW, HAUBRW, LLCLPLDA, LRLSLDA, and RWRlncD methods, the MSF-UBRW method achieves the highest AUC values under 5-fold CV and LOOCV. In addition, case studies of prostate cancer, ESCC, and NSCLC also confirm the prediction ability of the MSF-UBRW method.
Although the MSF-UBRW method has achieved good prediction results, it still have some limitations. Existing experimental data are inadequate, which limits the prediction performance of the MSF-UBRW method. In the future, as more LDA data are available, the MSF-UBRW method will be improved. However, the complexity and heterogeneity of biological data also bring some difficulties in improving the prediction ability of the algorithm. In the future, we will integrate data from different sources and improve the integrity and quality of experimental data to achieve higher prediction performance.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}