Abstract
Translation is a fundamental process in all living organisms that involves the decoding of genetic information in mRNA by ribosomes and translation factors. The dysregulation of mRNA translation is a common feature of tumorigenesis. Protein expression reflects the total outcome of multiple regulatory mechanisms that change the metabolism of mRNA pathways from synthesis to degradation. Accumulated evidence has clarified the role of an increasing amount of mRNA modifications at each phase of the pathway, resulting in translational output. Translation machinery is directly affected by mRNA modifications, influencing translation initiation, elongation, and termination or altering mRNA abundance and subcellular localization. In this review, we focus on the translation initiation factors associated with cancer as well as several important RNA modifications, for which we describe their association with cancer.
1. Introduction
Translation is an essential step in the regulation of gene expression. Cells consume a large amount of energy in the process of mRNA translation and protein synthesis [1]. In eukaryotes, translation initiation is a complex and highly regulated process, and this regulation is necessary for many important life processes, requiring the participation of at least a dozen protein factors [2]. At the same time, eukaryotic initiation factors (eIFs) regulate the translation of mRNA, which is crucial for gene expression. This regulation primarily occurs at the initiation stage of eukaryotic translation, which is the rate-limiting step of protein synthesis. The dysregulation of mRNA translation is a collective characteristic of tumorigenesis, and aberrant changes in the components of the translational machinery are also frequently found in cancer. For instance, the atypical behavior of the eIF complex activated by upstream signaling pathways has been detected in non-cancer-related human diseases and numerous tumors, resulting in the selective expression of the encoded proteins associated with tumorigenesis, metastasis, or anti-tumor drug resistance [3,4]. The translational regulation of pre-existing mRNA is more direct, faster, and more powerful than transcriptional regulation. It sensitively alters the levels of encoded proteins in cells, thereby changing the expression of the proteome rapidly without requiring alterations in RNA synthesis to allow cells to adapt to physiological and pathological conditions. The translation of eukaryotic genes is a very complex and precise process. The four main stages include initiation, elongation, termination, and the recycling of ribosomes [5].
Over the last five decades, RNA modifications have been identified using a variety of methods. However, it was only in the last decade that eukaryotic RNA modifications were identified and characterized, primarily in transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), followed by mRNAs and various noncoding RNAs [6]. The specific chemical modification of mRNA is an effective way to modulate molecular function, and the modification of mRNA and proteins can alter downstream signaling pathways. Therefore, in recent years, the study of RNA epitope transcriptomics has emerged as a promising new area in the field of RNA modification. The dysregulation of RNA modification pathways has also been found in human cancers, which suggests that they may be ideal targets for cancer treatment [7]. Global biological macromolecules (RNAs, DNAs, sugars, lipids, and proteins) undergo synthetic and covalent modifications. The widest variety of modifications has been found on proteins and RNA in animal cells. Over the past 50 years, more than one hundred different types of post-transcriptional RNA modifications have been identified using various methods [8]. With the deepening of research, it became clear that many molecular processes are regulated by precise RNA modifications, including RNA decay, splicing, localization, translation, stability, and metabolism, thus maintaining the diversity of genetic information. The epigenetic modification of RNA is mediated by three distinct classes of proteins that specialize in “writing” (catalyzing the deposition of specific modifications), “erasing” (catalyzing the removal of specific modifications), and “reading” (recognizing and binding specifically modified nucleotides), thereby affecting the fate of RNA [9]. The modification of RNA is a dynamic process in the body that allows cells to respond rapidly to environmental signals at any time. Adapting to a changing microenvironment, for instance, caused by stress or chemotherapeutic drug treatment, is essential for cancer cell development, which suggests that RNA modifications may play a key role in tumor progression [10,11]. Historically, cancer itself has also been a disease characterized by epigenetic or genetic alterations in tumor suppressor genes and oncogenes [12]. Although RNA modifications are sometimes not deemed to be cancer-initiating factors, numerous studies have illustrated that aberrant RNA modifications are often involved in functions affecting cell self-renewal, survival, proliferation, invasion, differentiation, and chemoresistance, all of which are associated with the characteristics of cancer [13,14,15].
In this review, we examine the present understanding of the fundamental function of eIFs in translation initiation, with a specific discussion of the dysregulation of eIFs and their roles in the initiation of mRNA translation as well as in tumorigenesis and metastasis (Figure 1). In addition, we describe the cellular and molecular functions of RNA modifications in regulating gene expression programs, focusing on N6-methyladenosine (m6A), N6,2′-O-dimethyladenosine (m6Am), 5-methylcytosine (m5C), pseudouridine (Ψ), and N4-acetylcytidine (ac4C) and their roles in cancer physiopathology (Figure 1). In addition, we provide insights into the mechanisms of these post-transcriptional modifications affecting tumor progression and maintenance.
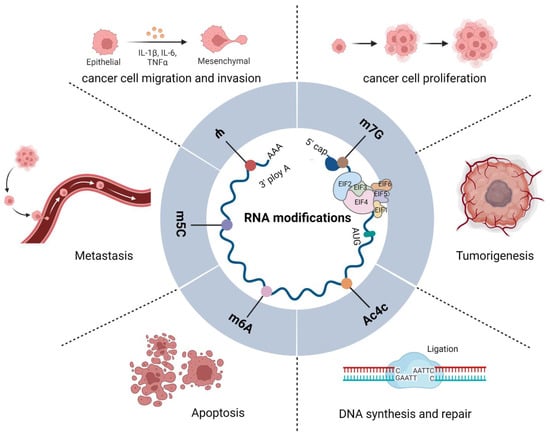
Figure 1.
Functions of eIFs and RNA modifications in tumors. Diagram showing the functions of eIFs and m6A, m6Am, m5C, Ψ, and ac4C in RNA modification. A variety of eIFs constitute translation initiation complexes that regulate RNA modification and cellular protein translation, directly or indirectly; they interact with one another to regulate the occurrence, development, and biological behavior of tumors.
2. Overview of eIFs in Translation Initiation
Translation initiation is a complex process during which eIFs assemble the initiator tRNA (Met-tRNAiMet) and the 40S and 60S ribosomal subunits into 80S ribosomes at the mRNA initiation codon. Among them, Met-tRNAiMet is located at the ribosomal P site of the initiation codon [16]. The complex initiation process of eIF-mediated 80S ribosome formation involves four stages [5] (Figure 2). First, eukaryotic initiation factor 2 (eIF2) selects Met-tRNAiMet from the extended tRNA pool and combines the eIF2/GTP/Met-tRNAiMet ternary complex (TC) and other eIFs with the 40S subunit to form a 43S preinitiation complex. Second, the eukaryotic initiation factor 4 subunit E (eIF4E) (cap binding) and F(eIF4F) initially recognize the N7-methylguanosine (m7G) cap at the 5′-terminus, allowing the 43S complex to bind to the mRNA. Then, the mRNA-bound ribosome complex moves along the 5′ untranslated region (5′UTR) from its initiation binding site to the initiation codon, forming a 48S initiation complex. The initiation codon pairs with the promoter anticodon of tRNA in this 48S initiation complex. Finally, the displacement of factors from the 48S complex and the joining of the 60S subunit to form an 80S ribosome leaves Met-tRNAiMet in the ribosomal P site. This facilitates the synthesis of the first peptide bond, and the process continues to the translational elongation stage. In eukaryotes, translation initiation culminates with formation of an 80S initiation complex in which Met-tRNAi Met is bound in the P site of the ribosome. Translation and translational regulation are considered key links in the adaptation of the tumor stress response to overcome various conditions, such as continuous proliferation, immune surveillance, the tumor microenvironment, and the cytotoxicity of drugs [17].
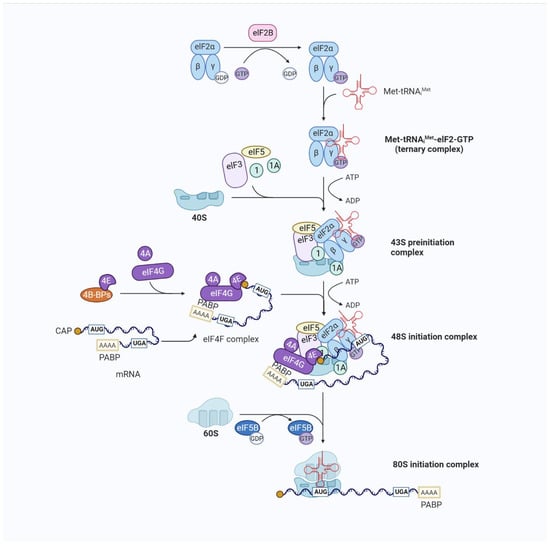
Figure 2.
Overview of eukaryotic translation initiation. Translation is an iterative process. The typical mechanism for the regulation of translation occurs at the rate-limiting stage of initiation, starting with the formation of the EIF2/GTP/Met-tRNAiMet TC and involving the control of the functional 40S subunit and its related factors (the 43S preinitiation complex (43S PIC)) assembly. The 43S PIC is a large complex formed by the binding of the 40S ribosomal subunit to the eukaryotic translation initiation factors eIF1, eIF1A, eIF3, and eIF5 and the TC. The recruitment of the 43S PIC to the mRNA template is facilitated by eIF4F, a complex consisting of the mRNA 5′-cap-binding subunit (eIF4E), a large scaffold protein (eIF4G), and an RNA helicase (eIF4A), resulting in the 48S PIC form. eIF4F recruits ribosomes to mRNA through the eIF4E-mRNA cap and eIF4G-eIF3 interaction to form the 48S initiation complex. eIF4G also interacts with poly(A)-binding protein (PABP), which binds to the mRNA 3′ poly(A) tail, resulting in mRNA circularization to stabilize mRNA and facilitate translation. The eIF4A helicase, which unfolds structures near the mRNA 5′-cap with other stronger helicases (DHX29), is involved in the initial interaction of eIF4F with the 5′ end of mRNA and can also facilitate the scanning of the 40S ribosomal subunit towards the start codon by resolving secondary structures in the 5′ untranslated region (UTR). The recognition of the initiation codon by the 43S PIC results in the release of eIF and the joining of the 60S subunit. The formation of the translationally competent 80S ribosome marks the end of initiation and the beginning of elongation.
Disorders in mRNA translation play a significant role in the pathogenesis of malignant tumors. Abnormal translation pathways promote tumor growth and cell transformation. In normal cells, aberrant translational pathways affect energy, stress, nutrient supply, gene expression, and ribosome production; however, in some cells they are overactivated and promote cancer [18,19]. Eukaryotes have evolved a complex translation initiation pathway. There are at least 12 dedicated proteins, the eIFs, that each play critical roles in the process, and several of these factors are comprised of multi-subunit proteins. A host of eIFs are protein complexes composed of a few subunits. The dysregulation of their expression through phosphorylation, downregulation, or overexpression leads to various types of carcinogenic progression [17,20,21,22]. Table 1 lists the functions of eIFs and their associated subunits in translation initiation.

Table 1.
Functions of eIFs and subunits in translation initiation. eIF—eukaryotic translation initiation factor; mRNA—messenger RNA; GTP—guanosine triphosphate; GDP—guanosine diphosphate; GAP—GTPase activating protein; tRNA—transfer RNA; TC—ternary complex; PIC—preinitiation complex; ATP—adenosine triphosphate; m7G—7-methylguanosine; UTR—untranslated region; PABP—poly(A)-binding protein; ATPase—adenosine triphosphatase; GTPase—guanosine triphosphatase; Met-tRNAiMet—initiator tRNA.
2.1. eIF1 and Tumors
Eukaryotic initiation factor 1 (eIF1) and eukaryotic initiation factor 1 subunit A (eIF1A) are essential to facilitate the initiation of the translational scanning process. Research has confirmed that when both eIF1 and eIF1A bind to 40S, the open conformation of the “latch” involving the mRNA entry channel undergoes structural rearrangement, allowing the PIC to scan. By contrast, the 40S/eIF1A complex may prevent scanning by leaving the “latch” in a locked conformation after eIF1 release. Importantly, eIF1 and eIF1A together stimulate the speed of TC binding to 40S and accurate AUG recognition; in the absence of eIF1, TC binds more tightly to the PIC, resulting in a sub-stable state of the TC that facilitates scanning but is incompatible with start codon recognition and binds to the “open” conformation of the PIC. Upon AUG recognition, the TC achieves a more stable interaction with the P-site by the isomerization reaction, which requires eIF1 dissociation and subsequent rearrangement to a closed 40S conformation [38,39]. Recently, it was discovered that somatic mutations in eIF1A, especially the N-terminal tail, are related to uveal cancer, thyroid cancer, and ovarian cancer [40,41,42,43].
Cancer-related N-terminal tail mutants reduce the interaction between eIF1A and the A-site ribosomal scanning arrest proteins Rps10 and Rps3 [44]. The presence of eIF1A mutations has also been reported in diverse types of melanoma and epithelial carcinoma [41,42]. However, further research is needed to reveal the biological functions of eIF1A in cells and how these functions contribute to cancer.
2.2. eIF2 and Tumors
eIF2 is a heterotrimeric initiation factor composed of eIF2α, β, and γ subunits, which together participate in the formation of the eIF2–Met-tRNAiMet–GTP complex. During initiation, the GTP in the TC is hydrolyzed to GDP, and eIF2–GDP must be recycled to eIF2–GTP for renewed TC assembly, a reaction catalyzed by the heteropentameric eIF2B complex, in particular the carboxy-terminal segment of eIF2Bε, which interacts directly with the G domain of eIF2γ and the lysine-rich regions of eIF2β. The other eIF2B subunits also contribute to eIF2–GDP binding through interactions with eIF2α; these interactions are enhanced by the phosphorylation of Ser51 by one of the eIF2α kinases, which are activated in stress conditions to downregulate general initiation. Meanwhile, the recycling of eIF2–GDP by eIF2B is negatively regulated by the formation of a competing eIF5–eIF2–GDP complex [45,46,47].
Currently, 29 eIF2B mutations have been reported, all of which render eIF2B insensitive to eIF2 phosphorylation [45]. In addition, the catalytic subunit of eIF2β is upregulated in diverse cancers and is related to tumorigenesis. The inhibition of this subunit in vitro can slow tumor progression; therefore, eIF2β may be an ideal therapeutic target for cancer [48]. One study found that the deletion of eIF2β led to G1 arrest in lung cancer cells, and the authors speculated that eIF2β knockout might have done so by reducing the binding of GTP to eIF2; however, more in-depth research is needed to support this hypothesis [48]. The translation of activating transcription factor 4 is promoted by phosphorylated eIF2α and is involved in osteoblast differentiation and bone formation [49,50]. Furthermore, eIF2α has been reported to be elevated in transformed lymphocytes from patients with non-Hodgkin’s lymphoma; this phenomenon was not observed in the mantle zones and surrounding paracortices, demonstrating that eIF2α expression was positively correlated with lymphocyte proliferation [51]. The aggressiveness of various lymphoma subtypes is positively linked to the expression level of eIF2 [52]. Collectively, eIF2 plays a significant role in protein synthesis, ribosomal energy metabolism, the cancer cell cycle, tumorigenesis, and development.
2.3. eIF3 and Tumors
eIF3 is the most complex and largest initiation factor in the translation initiation complex (TIC) with 13 subunits, eIF3a–m. They can be assembled into different eIF3 complexes. Among these subunits, eIF3a, b, c, e, f, and h are core components of the eIF3 complex [53]. eIF3 contributes to the hyperactivation of the translation initiation machinery and thereby plays a significant role in tumorigenesis [54]. The overexpression of eIF3 complex subunits, including eIF3a, b, c, h, and i, promotes the malignant transformation of fibroblasts by stimulating global protein synthesis and the translation of specific mRNA transcripts [55].
eIF3a, c, and i are overexpressed in a variety of cancers [56]. Ge Li et al. found that eIF3a not only promoted the expression of c-myc and cyclin D1 proteins but also blocked the phosphorylation of Raf-1 and ERK and affected the cell differentiation and morphological maturation of chronic myeloid leukemia cells [57]. eIF3c may serve as a direct target of YTHDF1, the “reader” of m6A. YTHDF1 enhances the translation of eIF3c in an m6A-dependent manner by binding to eIF3c mRNA, thereby promoting tumorigenesis and metastasis in ovarian cancer [58]. In addition, eIF3i upregulates COX-2 synthesis to regulate Wnt/β-catenin signaling and integrates with the PI3K/mTOR signaling pathway to induce abnormal colorectal cancer (CRC) proliferation and tumorigenesis [59]. According to molecular evidence, eIF3d may slow the progression of prostate cancer by regulating the translation of proteins involved in multiple signaling pathways, including those associated with the cell cycle and drug responses [60].
eIF3h is highly expressed in prostate and breast cancer. Interestingly, the expression of eIF3h was positively associated with poorly differentiated and aggressive prostate cancer [61]. Likewise, a high expression level of eIF3h was related to the migration, proliferation, and invasion of human hepatocellular carcinoma [62]. The amplification of the eIF3h gene was found in non-small cell lung cancer and CRC by fluorescence in situ hybridization and genome-wide analysis, respectively [63,64]. The eIF3-p48 gene was identified for the first time as a common integration site of murine mammary tumor virus in murine mammary tumors. The eIF3-p48 mutation observed in murine mammary tumor virus-induced tumors and hyperplasia contributes to the malignant transformation of mammary epithelial cells [65]. It has been reported that eIF3e expression was reduced in 31 percent of non-small-cell lung cancer patients and 37 percent of breast cancer patients. In contrast, the high expression of wild-type eIF3e does not promote malignant transformation, suggesting that wild-type eIF3e may have a tumor suppressor effect while truncated eIF3e has oncogenic potential [66].
eIF3f is the only eIF3 subunit that is downregulated in pancreatic cancer and melanoma. eIF3f induces apoptosis by inhibiting protein synthesis and proliferation in pancreatic cancer and melanoma cells, whereas eIF3f may act as a negative regulator of translational effects, and its low expression protects melanoma cells from apoptosis [67]. In fact, decreased eIF3f mRNA transcript levels are commonly detected in tumors such as breast cancer, ovarian cancer, melanoma, and pancreatic cancer [68]. Taken together, eIF3e and eIF3f may act as negative regulators of mRNA translation.
2.4. eIF4 and Tumors
The eIF4 complex has multiple components that can recruit ribosomal subunits to mRNA [69]. Mammalian eIF4B can stimulate the helicase activity of eIF4A, an activity it shares with its homolog, eIF4H. In addition, the binding and scanning of mRNA transcripts by the PIC in vitro is highly dependent on eIF4B [70,71]. Another study showed that eIF4B increased the efficiency with which eIF4G stimulated ATP hydrolysis coupled to RNA duplex unwinding by eIF4A, while eIF4H was less efficient in this regard. This is because eIF4H is shorter and lacks most of the carboxy-terminal region of eIF4B [72]. eIF4A plays an even more important role when it is mapped as a molecular nexus downstream of critical oncogenic signaling pathways (RAS, PI3K/AKT/mTOR, and MYC). It provides a direct link between the necessary steps in lymphomagenesis metastasis and the scanning of the 43S preinitiation complex [73,74].
At present, the function of eIF4B has not been fully elucidated. eIF4B binds to eIF4F to form a complex and regulates the activity of eIF4F, and it is considered unimportant for translation initiation [17]. Previous studies have shown that during translation initiation, eIF4B stimulates eIF4A activity by regulating the eIF4A conformational cycle and binding to the eIF3 complex through the eIF3A subunit [72,75]. In addition, eIF4B can be phosphorylated by activated S6K kinase, thereby enhancing the interaction between eIF4B and eIF3A, which is a downstream target of mTOR [76]. Targeting eIF4B suppresses the chemoresistant and metastatic properties of non-small cell lung cancer [77]. eIF4B has been shown to ensure cell survival by specifically promoting the translation of genes associated with cell proliferation as well as survival [78].
eIF4E is a cap-binding protein that specifically recognizes the 5′ m7G cap through stacking interactions with two tryptophan residues between two conserved tryptophans found in the cap-binding pocket of eIF4E. Meanwhile, eIF4E plays a key role in mRNA recruitment to control translation initiation and the translation rate [17]. In addition, eIF4E acts directly as an oncogene in vivo. Transgenic mice overexpressing eIF4E will experience increased tumorigenicity and resist treatment with drugs [79]. Numerous studies have elucidated that eIF4E and phosphorylated eIF4E are present in various types of cancer. The overexpression of eIF4E has been found in approximately 30% of human cancer cases [80,81]. MNK1 and MNK2 are recruited by the eIF4G protein of eIF4E to phosphorylate eIF4E, resulting in uncontrolled translation and the inhibition of apoptosis and proliferation [82,83]. eIF4E serves as the subunit of the eIF4F complex, and its ability to build a cap-binding eIF4F complex is regulated by 4E-binding protein 1 (4E-BP1). In addition, the phosphorylation of eIF4E contributes to its interaction with 4E-BP1 [84,85]. The activation of the mTOR pathway by the phosphorylation of 4E-BP1 reduces the binding of eIF4E to 4E-BP1, resulting in eIF4E hyperactivation [86].
When eIF4E interacts with the eIF4F complex, the hyperactivation of eIF4E results in translation initiation. Thus, the hyperphosphorylation of 4E-BP1 is connected with CRC [87], breast cancer [88], ovarian cancer [89], prostate cancer [90], lung cancer [91], lymphoma [92], and head and neck cancers [93]. In vivo, the high expression of eIF4E resulted in lung cancer, lymphoma, and angiosarcoma in transgenic mice [79]. Disrupting the interaction of eIF4E with the cap may be a possible anti-tumor approach, and researchers have conducted experiments to achieve this goal [94]. Likewise, in vitro studies reported that breast cancer cell migratory activity and invasiveness were significantly attenuated when eIF4E was knocked out [95].
eIF4G is a scaffold protein of the TIC, responsible for recruiting ribosomal subunits, mRNA, and cap-binding complexes to assemble in order to accomplish translation. eIF4G enhances the association of eIF4E with the cap structure through an allosteric enhancement mechanism [96]. eIF4G has binding sites for eIF4E [96], eIF4A [72], and eIF3a through an N-terminal poly(A)-binding protein (PABP) site [18,21]. Studies have shown that the inhibition of eIF4G/eIF4E complex interaction by the translation initiation inhibitor 4EGI-1 leads to growth restriction in human melanoma and breast cancer cells [97].
eIF4H is a cofactor of eIF4A. At the same time, eIF4H and eIF4B have similar abilities to stimulate eIF4A’s helicase activity; however, the stimulation by eIF4B is more pronounced in the presence of eIF4G. eIF4H and eIF4B bind to the same domain of eIF4A, and this binding is enhanced in the presence of ATP [98]. Studies have found that the dysregulation of eIF4A expression is associated with malignant transformation and is highly expressed in liver cancer and melanoma [99,100].
2.5. eIF5 and Tumors
eIF5 is one of the most important proteins in the translation initiation pathway and possesses two functions: at the N-terminus, it acts as a GTPase-activating protein (GTP hydrolyzation by eIF2) and an independent GDP dissociation inhibitor (through the controlled recycling of eIF2) [101,102]. At the same time, eIF5 may also inhibit the guanine nucleotide exchange factor eIF2B, which promotes eIF2 recycling [36]. eIF1 interacts with eIF5 and promotes the specific recognition of initiation codons. Histone deacetylase 2 can regulate the expression of eIF5 in lung cancer, and its high expression is associated with a poorer prognosis in lung cancer patients [103].
eIF5A is the gene encoding the eIF5 protein, which has extensive functions in translation elongation as well as termination; the deletion of eIF5A results in an overall translation elongation defect [104]. In mammals, eIF5 is the only known protein that undergoes post-translational hypusination, which is the incorporation of the rare amino acid hypusine [105]. The difference between eIF5A1 and eIF5A2 is that the former is expressed in all cells and is proportional to the rate of cell proliferation, while the latter is expressed in a tissue-specific manner and is almost undetectable in most cases [106]. Studies have shown that eIF5A2 increased resistance to doxorubicin by regulating the epithelial–mesenchymal transition of CRC. Thus, eIF5A2 may be an ideal potential target molecule for the treatment of CRC in the future [107].
eIF5B facilitates 60S ribosomal subunit ligation and can indirectly support the association of Met-tRNAiMet with the ribosome during translation initiation. eIF5B interacts with eIF5, eIF2A, and eIF1A and indirectly supports the binding of Met-tRNAiMet to ribosomes in translation initiation [108,109]. eIF5B is overexpressed in a variety of malignancies, and the abnormal expression of eIF5B is associated with glioblastoma [110], lung cancer [111], and hepatocellular carcinoma [112].
3. RNA Modifications
More recently, RNA modifications have been recognized as key post-transcriptional regulators of gene expression in eukaryotic cells. RNA modifications regulate multiple biological developmental pathways. The correct deposition of these modifications is essential for normal cellular development. Alterations in their deposition have been linked to several diseases, including cancer. Therefore, we concentrate on describing the presence of m6A, m6Am, m5C, Ψ, and ac4C and their pathophysiologic roles in tumors. In addition, we draw attention to recent insights into the mechanisms by which these post-transcriptional epigenetic modifications influence tumorigenesis and tumor progression.
3.1. m6A in Cancer
Among the many RNA modifications, the m6A modification is the most abundant and classical mRNA modification, which controls RNA translation, stability, and splicing. Simultaneously, m6A modification plays an important role in cell metabolism, differentiation, and growth. RNA can be dynamically and reversibly modified under the action of different types of enzymes; m6A interacts with “readers” (m6A-binding proteins), is catalyzed by “writers” (RNA methyltransferases), and is removed by “erasers” (demethylases) (Figure 3A). As more advanced techniques are employed in research, the profound molecular mechanisms of m6A RNA methyltransferases, demethylases, and binding proteins are beginning to be revealed. Furthermore, altered m6A RNA modification homeostasis has been associated with cancer [113]. The main roles of m6A writers, readers, and erasers in cancer are summarized in Table 2.
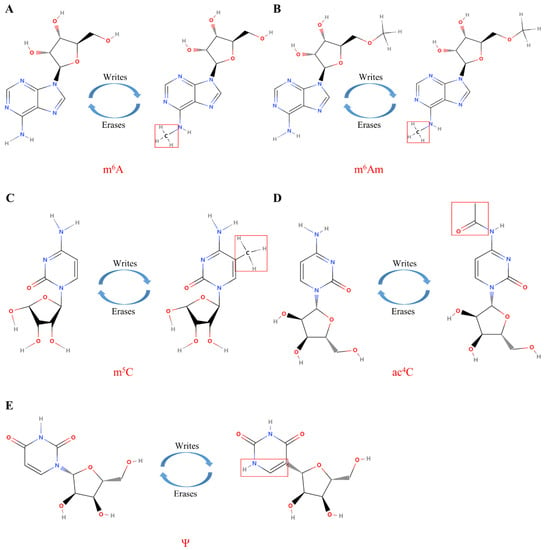
Figure 3.
The molecular composition of RNA modifications. (A) The m6A modification of RNA is mediated by proteins called “writes” and “erases”; (B) The m6Am modification of RNA is mediated by proteins called “writes” and “erases”; (C) The m5C modification of RNA is mediated by proteins called “writes” and “erases”; (D) The ac4C modification of RNA is mediated by proteins called “writes” and “erases”; (E) The Ψ modification of RNA is mediated by proteins called “writes” and “erases”. Under the catalysis of the corresponding enzymes, the molecular structure and the enrichment level of RNA are changed to play different biological roles.
Table 2.
Role of m6A in cancer. YTHDC1—YTH domain-containing 1; YTHDC2—YTH domain-containing 2; METTL3—methyltransferase-like 3; ALKBH5—alkB homolog 5; FTO—fat mass and obesity-associated gene; AML—acute myeloid leukemia; HCC—hepatocellular carcinoma; GSC—glioma stem cells.
3.1.1. m6A Writers in Cancer
A previous study showed that METTL3 was overexpressed in acute myeloid leukemia, in which it methylated BCL2, PTEN, and c-Myc mRNAs, leading to boosted translation and the inhibition of cell apoptosis and differentiation, thereby promoting leukemia progression [114]. In addition, another study confirmed that METTL3 could maintain acute myeloid leukemia cells in an undifferentiated state in vivo, thereby preventing the occurrence and development of myeloid leukemia [159]. In contrast, METTL14 acts as an oncogene in acute myeloid leukemia by augmenting the stability and translation of the MYB and c-Myc genes [115]. METTL3 promotes the translation of EGFR and TAZ, leading to lung and colon cancer cell growth, survival, and invasion [123]. METTL3 increases the methylation and stability of HBXIP mRNA by inhibiting the expression of the tumor suppressor gene let-7g in breast cancer, thereby inducing cell proliferation and survival [160]. Recent research studies have indicated that the abnormal accumulation of m6A has also been found in CRC. The aberrant deposition of m6A increases the expression of miRNA-1246, leading to the downregulation of the SPRED2 tumor suppressor gene and the induction of metastasis [118]. A more recent finding conveyed that METTL3 was highly expressed in prostate cancer and promoted cancer cell growth and invasion through SHH-GLI1 signaling [127]. Moreover, METTL3 was also found to control the expression of ITGB1, which affects ITGB1 binding to type I collagen and promotes bone metastasis in prostate cancer [128].
In hepatocellular carcinoma and glioblastoma, several reports have revealed the consistent oncogenic roles of METTL3 and METTL14 in these two cancer types. However, some reports have demonstrated the tumor-suppressing effects of both. Preliminary studies have shown that m6A methylation inhibits the self-renewal, progression, and tumorigenesis of glioblastoma stem cells by reducing the stability and expression of major ADAM19 oncogenic transcripts [12]. Meanwhile, another group of researchers found that METTL3 upregulation was observed to predict poorer patient survival and increased SOX2 mRNA expression and stability, promoting the growth of glioblastoma stem cells and shortening the survival time of mice [118]. Likewise, the m6A “writers” can facilitate or inhibit the occurrence and development of hepatocellular carcinoma. For example, METTL14 was originally considered as a tumor suppressor in liver cancer; METTL14 induced the increased expression of pri-miR-126, which inhibited tumor metastasis [161]. Conversely, Chen et al. discovered that METTL3 was upregulated in hepatocellular carcinoma patients and was correlated with worse prognosis due to its suppression of cytokine signaling-2 mRNA expression in an m6A-YTHDF2-dependent manner [121]. From the above reports, it can be concluded that the expression levels of m6A “writers”, “erasers”, and “readers” can account for the differences in m6A-targeted RNAs and in deposition, which lead to epigenetic differences. More detailed studies are needed, however, to characterize METTL3 and METTL14 in glioblastoma and hepatocellular carcinoma.
In endometrial cancer, 70% of tumors result in reduced m6A levels through the downregulation of METTL3 expression or METTL14 mutation. The loss of METTL3 and METTL14 complexes can upregulate the expression of AKT pathway members and lead to increased cell proliferation, which in turn exhibits tumor suppressor functions in endometrial cancer [162]. Likewise, the deletion of METTL3 in renal cancer promotes cell proliferation through the PI3K-AKT-mTOR pathway and activates the epithelial–mesenchymal transition pathway to promote cell invasion and migration [130].
Recently, some studies have been conducted on the role of METTL16 in cancer. In recent reports, changes in the expression, mutation, and loss of function of METTL16 all affected the occurrence and progression of CRC [163]. Other studies have reported that METTL16 is associated with the maturation of oncogene and tumor suppressor MALAT1 mRNA [164]. METTL16 regulates small nuclear RNA (snRNA) methylation and promotes tumor development by inducing changes in alternative splicing [165].
3.1.2. m6A Erasers in Cancer
The dysfunction of m6A erasers has been implicated in the development of cancer. In fact, fat mass and obesity-associated gene (FTO) nucleotide polymorphisms are associated with an increased risk of cancer and a variety of human diseases [166]. Likewise, the inhibition of FTO has been shown to prevent GSC growth, self-renewal, and tumorigenesis; FTO is associated with poor survival due to its regulation of ADAM19, a gene with key biological functions in glioblastoma stem cells [12]. FTO expression was also shown to be increased in other tumors. For example, in melanoma, FTO expression reduces the methylation of SOX10, CXCR4, and PD-1 by m6A in key tumor-promoting melanoma cell-intrinsic genes to promote tumorigenesis [143]. In cervical cancer, FTO interacts with E2F1 and MYC to increase its translation efficiency and promote cervical cancer cell migration and proliferation [140]. In lung cancer, FTO was shown to be related to poor prognosis due to its promotion of cell invasion and proliferation by controlling MZF1 expression and inhibition of lung cancer cell apoptosis [142]. Additionally, in breast cancer, the overexpression of FTO promotes cell proliferation and metastasis in vivo by downregulating BNIP3 expression [139].
ALKBH5, the second m6A demethylase, is overexpressed in several cancer types [146]. The expression of ALKBH5 is correlated with poor prognosis in glioblastoma and promotes the proliferation and tumor progression of glioblastoma stem cells by enhancing FOXM1 expression [147]. ALKBH5 enhances breast cancer stem cell stability and promotes tumorigenesis by reducing methylation in KLF4 and NANOG mRNA [167]. In gastric cancer, ALKBH5 boosts metastasis and invasion by demethylating the ncRNA NEAT1 [148]. Increased ALKBH5 in ovarian cancer predicts poorer survival, as the high expression of ALKBH5 activates the mTOR and BLC2-Beclin1 pathways to promote proliferation, invasion, and even autophagy [168]. Similar to other m6A receptors, ALKBH5 acts as a tumor suppressor in a variety of tumors. In pancreatic cancer cells, the deletion of ALKBH5 increases the methylation of the long noncoding RNA (lncRNA) KCNK15-AS1, which negatively regulates its expression and enhances cell migration. ALKBH5 also inhibits tumorigenesis by decreasing WAF-1 expression levels and hampering the activation of Wnt signaling [152].
3.1.3. m6A Readers in Cancer
In recent years, m6A “readers” have also been found to be abnormally expressed in tumors and associated with the development of cancer. Both YTH domain-containing 1 (YTHDC1) and YTHDF2 are overexpressed in CRC and are correlated with poor patient prognosis, metastatic potential, and cell proliferation. YTHDC2 controls the expression of the tumor-promoting gene HIF1a in these tumors [153]. The deletion of YTHDF1 inhibits the Wnt-β-catenin signaling pathway, thereby inhibiting tumorigenicity and tumor growth [154]. YTHDF1 is also upregulated in ovarian cancer. YTHDF1 upregulates the translation of eIF3c and promotes tumorigenesis and metastasis [58]. In contrast, YTHDF1 stimulates the translation of the tumor suppressor Hint2 in melanoma, thereby inhibiting tumor progression [155]. In acute myeloid leukemia, YTHDF2 can reduce the half-life of m6A-containing transcripts, thereby affecting tumor necrosis factor signaling and promoting apoptosis [156]. In addition, YTHDF2 was found to be abnormally overexpressed in lung cancer, where it promoted the translation of 6-phosphogluconate dehydrogenase to support tumor growth [158]. However, YTHDF2 was also able to inhibit cell proliferation and tumor growth and activate ERK and MEK signaling by inducing the degradation of EGFR mRNA in hepatoma cells [157].
3.2. m6Am Modifications in Cancer
m6Am was originally identified in viral mRNA and animal cells in the 1970s [169]. In contrast to m6A, which is an internal modification mainly present in the 3′ untranslated region (3′UTR), N6, 2′-O-dimethyladenosine (m6Am) contains one more methylated modification and is located adjacent to the cap structure of mRNA (Figure 3B). The first or second nucleotide near the m7G cap of the mRNA can be methylated at the 2′-hydroxyl. When the first nucleotide is 2′-O-methyladenosine (Am), it can be further methylated to m6Am at the N6 site under the catalysis of a specific methylase [170]. m6Am plays a crucial role in snRNA biogenesis [171], RNA splicing [172], mRNA cap-dependent translation, and stability [173,174].
Recent reports have shown that FTO, which is expressed at low levels in patient-derived cells, hinders the ability of cancer stem cells in CRC through its demethylase activity and increases m6Am levels in mRNA, resulting in enhanced tumorigenicity and chemoresistance in vivo [175] (Table 3). Past studies have acknowledged phosphorylated C-terminal domain-interacting factor 1 (PCIF1) as the only known m6Am “writer” protein; however, the additional catalytic functions of PCIF1 require further study [176]. The overexpression of m6Am methyltransferase PCIF1 in glioma blocks G0/G1 phase progression and induces glioma cell apoptosis, whereas the downregulation of PCIF1 promotes glioma cell proliferation [177]. Expression differences between normal tissues and tumors were obtained based on the Genotype Tissue Expression and Cancer Genome Atlas (TCGA) databases. It was found that the high expression of PCIF1 was negatively correlated with overall survival or disease-free survival in gastric cancer patients [178]. In addition, PCIF1 overexpression was found to promote the infiltration of CD4+ T cells in renal clear cell carcinoma and the infiltration of CD8+ T cells, macrophages, and B cells in thyroid cancer in vivo [179]. The role of m6Am in cancer development and progression has been newly proposed in recent years, and its specific mechanism remains to be elucidated by further research.
Table 3.
Role of m6Am in cancer. FTO—fat mass and obesity-associated gene; PCIF1—phosphorylated CTD-interacting factor 1.
3.3. 5-Methylcytosine
m5C is a universal and conserved RNA modification that exists in all areas of life (Figure 3C). m5C is widely present in a variety of RNA transcripts, including cytoplasmic and mitochondrial rRNA and tRNA, as well as mRNA, enhancer RNA, and some noncoding RNAs, but not in eukaryotic tRNAs. Among these, rRNA was found to hold the highest content [180]. In eukaryotic cells, the C5 methylation of RNA cytosines is catalyzed by the DNA methyltransferase homolog DNMT2 and the NOL1/NOP2/SUN domain (NSUN) family of enzymes. Recent research has identified the exact target nucleotides for the modification of the corresponding substrate proteins and RNA transcripts of various methyltransferases. These studies have provided a deeper molecular understanding of the oncogenic or tumor-suppressive effects caused by mutations in genes encoding m5C methyltransferases or by changes in the expression levels of these enzymes [181] (Table 4).
Table 4.
Role of the aberrant deposition of m5C in cancer. NSUN—NOP2/Sun; TET1—tet-eleven translocation 1; ALKBH1—alkB homolog 1; YBX1—Y-box binding protein 1; AML—acute myeloid leukemia; ALL—acute lymphoblastic leukemia; HCC—hepatocellular carcinoma.
3.3.1. m5C Writers in Cancer
In past studies, cytosine-5 methyltransferases have been revealed to play an important role in several cancers. The high expression of NOP2 was found to promote the proliferation of mouse fibroblasts [209]. At the molecular level, NOP2 interacts with the T-cell factor of the cyclin D1 promoter in cancer cells, recruiting telomerase RNA component elements and activating cyclin D1 transcription. It has been found that NOP2 is highly expressed in breast cancer, lung cancer, prostate cancer, and gallbladder cancer, and its expression level is positively correlated with poor prognosis [183,184,210]. However, whether NOP2 is involved in the methylation activity of rRNA needs to be further elucidated [211]. Changes in the expression of NSUN2 have also been associated with multiple tumor types, including head and neck squamous cell carcinomas and breast, bladder, skin, colon, ovarian, esophagus, gallbladder, and gastric cancers [182,186,196,197,198,199]. In one study, the high expression of NSUN2 was verified to regulate the fate of tumor suppressor and oncogene transcripts, thereby promoting proliferation [212]. Recently, NSUN2 was also shown to regulate metastasis and drug resistance through the methylation of lncRNA in esophageal cancer [192]. In another study, the NSUN2-mediated aberrant methylation of the 3′UTR of oncogenic mRNA transcripts, such as those of heparin-binding growth factor, augmented their stability via a link to YBX1 [186]. NSUN3 directly binds to hnRNPK and transcription factors in leukemia to increase chemotherapeutic drug resistance [185]. NSUN4 is associated with an increased risk of prostate, ovarian, and breast cancer, and its overexpression is associated with liver cancer [195,196]. Additionally, in glioblastomas, the expression of NSUN5 is correlated with poor survival [213], while in low-grade gliomas, the high expression of NSUN7 is associated with a shorter survival time [197]. However, it is unclear whether the underlying molecular mechanisms of NSUN2 in other cancers can regulate mRNA modification.
Regarding tRNA methylation, it has been shown that NSUN2 is upregulated in skin tumor cell populations depending on the suitable deposition of m5C on tRNAs [174]. The loss of NSUN2 leads to the hypomethylation of tRNA, resulting in the accumulation of 5′-tRNA fragments [14,214]. Indeed, it is increasingly recognized that the biogenesis of tRNA fragments is mainly stimulated by environmental stressors, and their aberrant expression in cancer suggests a significant function in tumorigenesis [215]. Little is known about their functions, and studies have revealed that 5′-tRNAs can regulate protein translation in response to stress by controlling the binding of various translation initiation factors to the TIC [216,217]. Importantly, the lack of NSUN2 leaves cells in an undifferentiated and proliferation-inhibited state, which is required for tissue or cancer stem cell self-renewal [188,214,218]. Other studies have shown that cancer cells require the precise regulation of tRNA methylation, protein synthesis, and the biological function of tRNA fragments to maintain cellular responses and tumor volume [219]. Alterations in rRNA methylation are also associated with tumors. The expression level of NSUN5 is correlated with poor survival in glioblastoma patients. The epigenetic deletion of NSUN5 in glioma downregulates rRNA methylation levels and subsequently increases growth factor translation, making glioma cells resistant to the stress-related enzyme NAD(P)H quinone dehydrogenase 1 sensitive [213]. Thus, these findings underscore the importance of intracellular tRNA and rRNA methylation for the synthesis of various proteins and also suggest a role of RNA methylases in cancer development and progression.
3.3.2. m5C Erasers in Cancer
Erasers of 5-hydroxymethylation have also been found to be altered in cancer. Mutations in or the altered expression of TET and ALKBH1 have been connected to certain malignancies. For instance, ALKBH1 and TET2 mutations are associated with myeloid and lymphoblastic leukemias [220,221], while TET1 is highly expressed in glioblastoma [222]. To the contrary, TET2 is downregulated in gliomas [200], whereas the epigenetic inhibition of TET3 may alter glioblastoma tumorigenesis [202]. While the mechanistic reasons are always related to DNA hydroxymethylation or demethylation deficiency, ALKBH1 and TET have also been indicated to oxidize m5C in RNA, suggesting that RNA hydroxymethylation deficiency is also associated with cancer.
3.4. Pseudouridine (Ψ)
Ψ, the C5 glycosidic isomer of uridine, was the first epigenetic modification to be discovered and is also one of the most abundant in RNA [223] (Figure 3E). Although Ψ was discovered decades ago, it was only recently that research began to reveal its biological functions (Table 5). Ψ was originally identified on tRNA and rRNA in yeast [224]; however, it has also been identified on different types of RNA, including small Cajal body-specific RNA, snRNA, tRNA, rRNA, miRNA, and lncRNA [225]. Most importantly, with current technological advances, further genome-wide analyses have successfully identified novel substrate mRNA molecules. The molecular function of Ψ in cancer is also becoming increasingly understood. The function of Ψ is mainly achieved through two important and different pathways: RNA-independent and RNA-dependent Ψ action. The RNA-dependent mechanism is mediated by an RNA–protein complex of small ribonucleoproteins, which consists of a box H/ACA snoRNA and four core proteins: glycine-arginine rich protein 1, nucleolar protein 10, nonhistone protein 2, and dyskerin (also known as NAP57 or DKC1) [226]. RNA-independent Ψ is catalyzed by a single pseudouridine synthase (PUS), which does not require RNA template strands for substrate recognition and catalysis [227]. In eukaryotic cells, there are more than 14 different Ψ enzymes, 10 of which are members of the PUS family [228]. Each of them has specific catalytic targets, but they also share some common targets.
Table 5.
Role of pseudouridylases in cancer. DKC1—dyskerin pseudouridine synthase 1; PUS1—pseudouridylase synthase 1; AML—acute myeloid leukemia; CLL—chronic lymphocytic leukemia; HCC—hepatocellular carcinoma.
3.4.1. RNA-Dependent Ψ Synthetases in Cancer
The main modified substrates of dyskerin are SnRNAs and rRNAs, which are also involved in the activation of the telomerase complex [223]. DKC1-inactivating mutations are associated with X-linked dyskeratosis congenita (X-DC) and thereby increase the risk of cancer development in patients with X-DC [244].
DKC1 gene mutations have been linked to various cancers, such as skin cancer [245], breast cancer [229], CRC [232], lung cancer [232], prostate cancer [237], head and neck cancers [234], glioma [233], and liver cancer [235], as well as hematological malignancies and bone marrow failure syndromes, including chronic lymphocytic leukemia and multiple myeloma [236,237]. Mechanistically, the downregulation of pseudoribosylated rRNA attenuates the internal ribosome entry-dependent translation of tumor suppressor genes, such as p27, p53, and suppressors of apoptosis (XIAP, BclXL) [238,246]. The enhanced expression of vascular endothelial growth factor is related to the knockdown of DKC1, leading to an increased risk of oncogenesis in cells [247]. Other recent studies have reported that the loss of snoRNA, which is modified by pseudouridine at specific rRNA residues, increases protein mistranslation in hepatocellular carcinoma cells, resulting in changes in the ribosomal elongation rate, tRNA selection efficiency, and translation efficiency and thus affecting cancer cell survival [248]. Another study of rRNA Ψ modification found a reduction in 1-methyl-3-α-amino-α-carboxyl-propyl pseudouridine (m1acp3Ψ) modification in CRC, which affected the normal interactions between ribosomal P-sites and tRNA, altering and attenuating the translation efficiency in cancer cells [249].
Conversely, other studies have suggested that dyskerin plays a carcinogenic role. For example, DKC1 expression levels, the pseudouridylation of rRNA, and reduced telomere length are associated with a better prognosis in breast cancer [250]. In gliomas, the high expression of DKC1 stimulates the increased expression of glioma growth regulators to promote glioma cell migration and progression [233]. The data thus far suggest that alterations in DKC1 expression or activity are strongly associated with cancer, but the exact mechanism may depend on the cell, tissue, or DKC1 substrate. Further studies are needed to explore whether DKC1 has oncogenic or tumor-suppressive effects.
3.4.2. RNA-Independent Ψ Synthetases in Cancer
The function of DKC1 in the occurrence and development of cancer is more well-studied, but little is known about the function of other pseudouridase enzymes, and only a handful of studies have shown their activity or expression related to tumors. For example, studies have identified PUS1 as a co-activator of retinoic acid receptor γ and other nuclear receptors in melanoma cells and breast cancer that simultaneously catalyze uridine in noncoding RNAs to pseudouridine to ensure proper protein folding and function [239]. PUS10 is present only in eukaryotes, and the dysregulation of PUS10 induces apoptosis in prostate cancer cells by reducing their sensitivity to the tumor necrosis factor-related apoptosis-inducing ligand [242]. Genomic alterations in PUS10 are pointedly correlated with lung cancer risk, but further studies are needed to demonstrate whether they are dependent on pseudouridase activity [243]. PUS7 is underexpressed in myelodysplastic syndromes, which may ultimately lead to a high risk of the disease transforming into acute myeloid leukemia [240]. Mechanistically, one study demonstrated the deletion of PUS7 in human hematopoietic stem and progenitor cells and that embryonic stem cells reduced PUS7-mediated pseudouridylation in a specific class of tRNA-derived RNA fragments, resulting in significantly higher rates of protein synthesis and leading to severe differentiation and growth defects [241]. Studies using patient-derived cancers and in vivo model cells will help uncover the clinical implications of targeting tRNA fragments for PUS7 deletion or aberration. Thus, the important role of tRNA fragments in reprogramming translation has been demonstrated [251].
3.5. ac4C in Cancer
ac4C, a recently acknowledged epigenetic modification within mRNA (Figure 3D), has been described as a crucial regulator of mRNA translation efficiency and stability. NAT10, the only known “writer” protein of ac4C, is thought to have vital effects on tumor metastasis and tumorigenesis. ac4C is present in more than 4000 human transcriptome regions. In human cervical cancer cells, ac4C is mainly enriched in the coding sequence region and decreases sequentially from the 5′UTR to the 3′UTR region of the gene transcript [252]. The function and mechanism of ac4C in gene expression regulation in a variety of cancers is summarized in Table 6. The downregulation of NAT10 was shown to result in the reduced ac4C modification of mRNA in specific regions of bladder cancer cells, impairing the translational stability and efficiency of BCL9L, SOX4, and AKT1. The deletion of NAT10 was also shown to reduce tumor burden in bladder cancer xenograft and transgenic mouse models [253]. Another study found that ac4C was elevated in the urine of CRC patients and was positively correlated with Duke’s CRC stage but significantly decreased after the radical resection of CRC [254]. Similarly, ac4C is also highly expressed in liver cancer patients. High ac4C expression in patients is correlated with an advanced tumor stage, high tumor stemness, high TP53 mutation rate, low stromal score, high immune score, and greater activation of the pathways of the DNA repair system and regulation of T-cell infiltration [255]. Other researchers have found that NAT10 plays a vital function in gastric cancer metastasis by regulating the writing pathway of the gastric cancer mRNA ac4C [256]. At the same time, some scholars have analyzed the comprehensive TCGA and Genotype Tissue Expression databases and found that the overexpression of NAT10 in multiple cancers is significantly associated with a poor prognosis when compared to normal tissues. Notably, NAT10 expression in liver cancer was positively correlated with immune infiltration, including CD4+ T cells, CD8+ T cells, B cells, etc. [257]. As mentioned above, ac4C plays a significant role in the development and occurrence of various cancers; however, as far as the current research is concerned, most of these studies focus only on clinical patients and data analysis. To date, only a few molecular mechanism experiments have been performed to elucidate the role of ac4C in the specific mechanisms of various cancers.
Table 6.
Role of ac4C in cancer. HNSCC—head and neck squamous cell carcinoma; KRPCC—kidney renal papillary cell carcinoma.
3.6. mRNA Modification and Translation Factors: Crosstalk between m6A and eIFs in Cancer
Interestingly, recent findings have shown a correlation between m6A modification and human translation initiation factors that regulate the development and occurrence of cancer. METTL3 interacts with eIF3h to promote translation by affecting its multimer conformation. METTL3 promotes oncogene translation and tumorigenesis through an mRNA loop mechanism; however, the deletion of eIF3h abolishes these functions. This ability is similar to eIF4G-PABPC1-mediated mRNA looping [62,63]. Studies have also reported that eIF4A/4B can target 50 Wilms’ tumor-associated protein (WTAP), an adaptor of the m6A RNA methyltransferase complex, and mRNA transcripts whose translation can be controlled by mTORC1 downstream kinase S6K promotion, thereby promoting the occurrence and development of tumors [258,259].
Additionally, the knockdown of METTL3 was shown to significantly inhibit the expression of translation initiation factors, including eIF (2B3, 3b, 3c, 3d, 4A1, and 5/5A), thereby inhibiting lung adenocarcinoma proliferation and reducing the invasive ability of cancer cells. It was also shown that m6A methylation is required during translation [260]. In vitro experiments have shown that the eIF3d gene inhibits prostate cancer progression by regulating translation, the cell cycle, drug response, and multiple signaling pathways at m6A levels [60]. After METTL3 knockout, the distribution of the translation initiation factor eIF3 core subunit eIF3b, the cytosolic m7G cap-binding protein eIF4e, and the nuclear m7G cap-binding protein CBP80 shift from heavier multimeric components to lighter submultimeric fractions, resulting in attenuated translation and inhibiting cancer cell survival, growth, and invasion [123].
Studies have elucidated that eIF4A3 upregulates the binding of circular RNA (circRNA) and WTAP, promoting the formation of WTAP/METTL3/METTL14 m6A methyltransferase in bladder cancer tissues and cell lines. The formation of complexes reduces the chemosensitivity of bladder cancer to chemotherapeutic agents [261]. METTL14 directly binds to eIF4G1 mRNA and reduces the stability of eIF4G1 RNA by mediating m6A modification, thereby regulating autophagy levels and effectively reducing the growth of oral squamous cell carcinoma [262]. METTL16 is an RNA methyltransferase primarily responsible for the deposition of N6-methyladenosine (m6A) in transcripts. In the cytosol, METTL16 directly interacts with eIF3a and b and rRNA through its Mtase domain to promote the assembly of the TIC. METTL16 is critical for hepatocellular carcinoma tumorigenesis [263].
Importantly, one study found that circRNAs are enriched in m6A motifs and drive translation under the action of eIF4G2 and the m6A reader YTHDF3 [264]. METTL3 triggers m6A mRNA methylation, which further regulates the MALAT1-miR-1914-3p-YAP axis by recruiting YTHDF1/3 and eIF3b to the TIC, thus increasing YAP mRNA translation and stability. The high expression of YAP enhances the induction of drug resistance and metastasis in non-small cell lung cancer [15]. Meanwhile, YTHDF1 negatively affects eIF3a and b through the m6A mechanism and reduces the proliferation and clone formation of Merkel cell carcinoma in vitro [265]. In another study, TCGA analysis identified that the m6A-regulating genes YTHDF1 and eIF3b were upregulated in CRC and confirmed that they were significantly differentially expressed between CRC and normal tissues by immunohistochemistry [266]. YTHDF1 promotes ovarian cancer tumorigenesis and metastasis by binding to m6A-modified eIF3c mRNA and enhancing eIF3c translation in a m6A-dependent manner while promoting overall translational output [264]. eIF2α kinase 2 bridges YTHDF3 and eIF3a to recognize the 5′UTR of m6A-methylated RNA and enhances YTHDF3/eIF3a complex stability in oxaliplatin-resistant CRC cells [267]. Following FTO silencing, YTHDF2 captures m6A-containing eIF4G1 transcripts, resulting in mRNA degradation and reduced eIF4G1 protein expression, thereby promoting autophagy and reducing tumorigenesis [268]. Emerging studies have reported that circPDE5A blocks the WTAP-dependent m6A methylation of eIF3c mRNA by forming a circPDE5A-WTAP complex, ultimately disrupting the translation of eIF3c for prostate cancer cell migration and invasion [269]. circPVRL3 was also predicted to bind eIF4A3 based on the structure of the internal ribosome entry, m6A modification, and open reading frame. The downregulation of circPVRL3 can promote the proliferation of gastric cancer [270].
4. Conclusions and Perspectives
In conclusion, numerous studies have demonstrated the importance of various RNA modifications and translational regulatory mechanisms in cancer. RNA modification and translation initiation are critical for tumor cell survival and proliferation and the expression of oncogenic proteins, affecting tumorigenesis and progression. RNA modification is a crucial post-transcriptional regulator of the gene expression program, and translation initiation is the critical and rate-limiting step; both have a key impact on the translational control of gene expression. At the same time, translation (translation rate, translation inhibition or enhancement, translation abnormality, etc.) can be determined by translation initiation factors as well as different modifications at different sites on the RNA transcripts. Recently, an increasing number of studies have focused on the functional network of the epitranscriptome and translational control via metabolism, epigenetics, and chromatin remodeling, and have recognized that the abnormal deposition of RNA modifications in cancer cells leads to mistranslation and a wide range of functional alterations.
Many enzymes responsible for RNA modification and regulation are targets of current cancer treatments. RNA epitranscriptomics, the exploration of RNA modifications, is a popular frontier. With the development of technology, new RNA modifications (such as ac4C) have been identified in cancer. Their biological functions and connections with cancer have been described, which has also provided new insights into future research. Identifying more unknown RNA-modifying enzymes and RNA modifications and determining the oncogenic or tumor-suppressive effects of aberrant modifications will guide the search for precise molecular targets and provide a theoretical basis for developing effective therapies against special tumor classes or other complex diseases. However, there is still much work to be completed to fully understand the complex relationship between RNA modification and translation in the development of tumor-targeted therapeutic drugs and biomarkers. At present, studies on translation regulation and RNA modifications are only the tip of the iceberg, but their rapid development will comprehensively expand our understanding of cancer.
Author Contributions
L.Z., Y.Z. (Yaguang Zhang) and S.Z.: writing—original draft (equal); writing—review and editing (supporting). L.Q., Y.Z. (Ying Zhou) and Y.Z. (Yang Zhang): writing—review and editing (supporting). J.X.: project administration (supporting); writing—review and editing (supporting). J.H.: conceptualization (lead); funding acquisition (lead); project administration (lead); writing—review and editing (lead). All authors have read and agreed to the published version of the manuscript.
Funding
This review was funded by the Science and Technology Plan Project of Sichuan Province, grant number 2021YJ0170; the National Natural Science Foundation of China (82203447), Postdoctoral Research Project, West China Hospital, Sichuan University (18HXBH068), and the Natural Science Foundation of Sichuan, China (2022NSFSC1424).
Institutional Review Board Statement
Not applicated.
Informed Consent Statement
Not applicated.
Data Availability Statement
All data are contained within this article and available from the corresponding author on reasonable request.
Conflicts of Interest
All authors declare no conflict of interest.
References
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312 Pt 1, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Lasko, P. Translational control in cellular and developmental processes. Nat. Rev. Genet. 2012, 13, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Yu, J.; Ward, R.; Liu, Y.; Hao, Q.; An, S.; Xu, T. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun. Signal. 2020, 18, 175. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Khoutorsky, A.; Mathews, M.B.; Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 791–807. [Google Scholar] [CrossRef]
- Verma, M.; Choi, J.; Cottrell, K.A.; Lavagnino, Z.; Thomas, E.N.; Pavlovic-Djuranovic, S.; Szczesny, P.; Piston, D.W.; Zaher, H.S.; Puglisi, J.D.; et al. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 2019, 10, 5774. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Li, S.; Mason, C.E. The Pivotal Regulatory Landscape of RNA Modifications. Annu. Rev. Genom. Hum. Genet. 2014, 15, 127–150. [Google Scholar] [CrossRef]
- Davalos, V.; Blanco, S.; Esteller, M. SnapShot: Messenger RNA Modifications. Cell 2018, 174, 498–498.e1. [Google Scholar] [CrossRef]
- Gkatza, N.A.; Castro, C.; Harvey, R.F.; Heiß, M.; Popis, M.C.; Blanco, S.; Bornelöv, S.; Sajini, A.A.; Gleeson, J.G.; Griffin, J.L.; et al. Cytosine-5 RNA methylation links protein synthesis to cell metabolism. PLoS Biol. 2019, 17, e3000297. [Google Scholar] [CrossRef]
- Chan, C.T.; Dyavaiah, M.; DeMott, M.S.; Taghizadeh, K.; Dedon, P.C.; Begley, T.J. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010, 6, e1001247. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.-G.; et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef]
- Jin, D.; Guo, J.; Wu, Y.; Du, J.; Yang, L.; Wang, X.; Di, W.; Hu, B.; An, J.; Kong, L.; et al. m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MA-LAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J. Hematol. Oncol. 2019, 12, 135. [Google Scholar] [CrossRef]
- Pestova, T.V.; Kolupaeva, V.G.; Lomakin, I.B.; Pilipenko, E.V.; Shatsky, I.N.; Agol, V.I.; Hellen, C.U.T. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 7029–7036. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.U.; Ur Rahman, M.S.; Jia, Z.; Jiang, C. Eukaryotic translation initiation factors and cancer. Tumor Biol. 2017, 39, 1010428317709805. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Liang, S.; Zhou, Y.; Chen, Y.; Ke, G.; Wen, H.; Wu, X. Decreased Expression of EIF4A1 After Preoperative Brachytherapy Predicts Better Tumor-Specific Survival in Cervical Cancer. Int. J. Gynecol. Cancer 2014, 24, 908–915. [Google Scholar] [CrossRef]
- Heikkinen, T.; Korpela, T.; Fagerholm, R.; Khan, S.; Aittomäki, K.; Heikkilä, P.; Blomqvist, C.; Carpén, O.; Nevanlinna, H. Eukaryotic translation initiation factor 4E (eIF4E) expression is associated with breast cancer tumor phenotype and predicts survival after anthracycline chemotherapy treatment. Breast Cancer Res. Treat. 2013, 141, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.J.; Van Meter, A.J.; Gadhikar, M.A.; Henderson, Y.C.; Yao, H.; Pickering, C.C.; Williams, M.D.; El-Naggar, A.K.; Sandulache, V.; Tarco, E.; et al. Phosphoproteomic analysis of signaling pathways in head and neck squamous cell carcinoma patient samples. Am. J. Pathol. 2011, 178, 548–571. [Google Scholar] [CrossRef] [PubMed]
- Sokabe, M.; Fraser, C.S. Human eukaryotic initiation factor 2 (eIF2)-GTP-Met-tRNAi ternary complex and eIF3 stabilize the 43 S preinitiation complex. J. Biol. Chem. 2014, 289, 31827–31836. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Clayton, J.; Shalev, A.; Hinnebusch, A.G. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000, 14, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Lin, K.Y.; Edmonds, K.A.; Yu, J.; Nadkarni, D.; Marintcheva, B.; Marintchev, A. eIF1A/eIF5B interaction network and its functions in translation initiation complex assembly and remodeling. Nucleic Acids Res. 2016, 44, 7441–7456. [Google Scholar] [CrossRef]
- Acker, M.G.; Shin, B.S.; Dever, T.E.; Lorsch, J.R. Interaction between eukaryotic initiation factors 1A and 5B is required for efficient ribosomal subunit joining. J. Biol. Chem. 2006, 281, 8469–8475. [Google Scholar] [CrossRef]
- Beilsten-Edmands, V.; Gordiyenko, Y.; Kung, J.C.; Mohammed, S.; Schmidt, C.; Robinson, C.V. eIF2 interactions with initiator tRNA and eIF2B are regulated by post-translational modifications and conformational dynamics. Cell Discov. 2015, 1, 15020. [Google Scholar] [CrossRef]
- Kashiwagi, K.; Takahashi, M.; Nishimoto, M.; Hiyama, T.B.; Higo, T.; Umehara, T.; Sakamoto, K.; Ito, T.; Yokoyama, S. Crystal structure of eukaryotic translation initiation factor 2B. Nature 2016, 531, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.; Do, A.; Hershey, J.W.; Fraser, C.S. Human eukaryotic initiation factor 4G (eIF4G) protein binds to eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J. Biol. Chem. 2013, 288, 32932–32940. [Google Scholar] [CrossRef]
- LeFebvre, A.K.; Korneeva, N.L.; Trutschl, M.; Cvek, U.; Duzan, R.D.; Bradley, C.A.; Hershey, J.W.; Rhoads, R.E. Translation initiation factor eIF4G-1 binds to eIF3 through the eIF3e subunit. J. Biol. Chem. 2006, 281, 22917–22932. [Google Scholar] [CrossRef]
- Valásek, L.; Nielsen, K.H.; Zhang, F.; Fekete, C.A.; Hinnebusch, A.G. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 2004, 24, 9437–9455. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, Q.; Miller, W.A.; Goss, D.J. Eukaryotic translation initiation factor 4G (eIF4G) coordinates interactions with eIF4A, eIF4B, and eIF4E in binding and translation of the barley yellow dwarf virus 3′ cap-independent translation element (BTE). J. Biol. Chem. 2017, 292, 5921–5931. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, E.; Jenni, S.; Kabha, E.; Takrouri, K.J.; Yi, T.; Salvi, N.; Luna, R.E.; Gavathiotis, E.; Mahalingam, P.; Arthanari, H.; et al. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc. Natl. Acad. Sci. USA 2014, 111, E3187–E3195. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, L.; Imataka, H.; Pelletier, J. Cap-dependent eukaryotic initiation factor-mRNA interactions probed by cross-linking. RNA 2008, 14, 960–969. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Korneeva, N.L.; Song, A.; Gram, H.; Edens, M.A.; Rhoads, R.E. Inhibition of Mitogen-activated Protein Kinase (MAPK)-interacting Kinase (MNK) Preferentially Affects Translation of mRNAs Containing Both a 5′-Terminal Cap and Hairpin. J. Biol. Chem. 2016, 291, 3455–3467. [Google Scholar] [CrossRef]
- Sun, Y.; Atas, E.; Lindqvist, L.; Sonenberg, N.; Pelletier, J.; Meller, A. The eukaryotic initiation factor eIF4H facilitates loop-binding, repetitive RNA unwinding by the eIF4A DEAD-box helicase. Nucleic Acids Res. 2012, 40, 6199–6207. [Google Scholar] [CrossRef]
- Jennings, M.D.; Zhou, Y.; Mohammad-Qureshi, S.S.; Bennett, D.; Pavitt, G.D. eIF2B promotes eIF5 dissociation from eIF2*GDP to facilitate guanine nucleotide exchange for translation initiation. Genes Dev. 2013, 27, 2696–2707. [Google Scholar] [CrossRef]
- Singh, C.R.; Lee, B.; Udagawa, T.; Mohammad-Qureshi, S.S.; Yamamoto, Y.; Pavitt, G.D.; Asano, K. An eIF5/eIF2 complex antagonizes guanine nucleotide exchange by eIF2B during translation initiation. EMBO J. 2006, 25, 4537–4546. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. The Scanning Mechanism of Eukaryotic Translation Initiation. Annu. Rev. Biochem. 2014, 83, 779–812. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Lorsch, J.R. The Mechanism of Eukaryotic Translation Initiation: New Insights and Challenges. Cold Spring Harb. Perspect. Biol. 2012, 4, a011544. [Google Scholar] [CrossRef]
- Ewens, K.G.; Kanetsky, P.A.; Richards-Yutz, J.; Purrazzella, J.; Shields, C.L.; Ganguly, T.; Ganguly, A. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- Karunamurthy, A.; Panebianco, F.; Hsiao, S.J.; Vorhauer, J.; Nikiforova, M.N.; Chiosea, S.; Nikiforov, Y.E. Prevalence and phenotypic correlations of EIF1AX mutations in thyroid nodules. Endocr.-Relat. Cancer 2016, 23, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Etemadmoghadam, D.; Azar, W.J.; Lei, Y.; Moujaber, T.; Garsed, D.W.; Kennedy, C.J.; Fereday, S.; Mitchell, C.; Chiew, Y.-E.; Hendley, J.; et al. EIF1AX and NRAS Mutations Co-occur and Cooperate in Low-Grade Serous Ovarian Carcinomas. Cancer Res. 2017, 77, 4268–4278. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.-E.; Rowley, S.M.; Doyle, M.A.; Li, J.; Gilks, C.B.; Moss, P.; et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget 2015, 6, 37663–37677. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, U.; Koning, F.; Ashkenazi, S.; Stelzer, G.; Leshkowitz, D.; Dikstein, R. Cancer-Associated Eukaryotic Translation Initiation Factor 1A Mutants Impair Rps3 and Rps10 Binding and Enhance Scanning of Cell Cycle Genes. Mol. Cell. Biol. 2019, 39, e00441-18. [Google Scholar] [CrossRef] [PubMed]
- Pavitt, G.D.; Yang, W.; Hinnebusch, A.G. Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol. Cell. Biol. 1997, 17, 1298–1313. [Google Scholar] [CrossRef]
- Sudhakar, A.; Ramachandran, A.; Ghosh, S.; Hasnain, S.E.; Kaufman, R.J.; Ramaiah, K.V. Phosphorylation of serine 51 in initiation factor 2 alpha (eIF2 alpha) promotes complex formation between eIF2 alpha (P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 2000, 39, 12929–12938. [Google Scholar] [CrossRef]
- Krishnamoorthy, J.; Mounir, Z.; Raven, J.; Koromilas, A. The eIF2α kinases inhibit vesicular stomatitis virus replication independently of eIF2 phosphorylation. Cell Cycle 2008, 7, 2346–2351. [Google Scholar] [CrossRef]
- Saito, A.; Ochiai, K.; Kondo, S.; Tsumagari, K.; Murakami, T.; Cavener, D.R.; Imaizumi, K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem. 2011, 286, 4809–4818. [Google Scholar] [CrossRef]
- Donzé, O.; Jagus, R.; Koromilas, A.E.; Hershey, J.W.; Sonenberg, N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995, 14, 3828–3834. [Google Scholar] [CrossRef]
- Wang, S.; Rosenwald, I.B.; Hutzler, M.J.; Pihan, G.A.; Savas, L.; Chen, J.J.; Woda, B.A. Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-Hodgkin’s lymphomas. Am. J. Pathol. 1999, 155, 247–255. [Google Scholar] [CrossRef]
- Lam, N.; Sandberg, M.L.; Sugden, B. High physiological levels of LMP1 result in phosphorylation of eIF2 alpha in Epstein-Barr virus-infected cells. J. Virol. 2004, 78, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Spilka, R.; Ernst, C.; Mehta, A.K.; Haybaeck, J. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013, 340, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.Y.; Kranzusch, P.J.; Cate, J.H.D. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 2015, 522, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pan, X.; Hershey, J.W. Individual Overexpression of Five Subunits of Human Translation Initiation Factor eIF3 Promotes Malignant Transformation of Immortal Fibroblast Cells. J. Biol. Chem. 2007, 282, 5790–5800. [Google Scholar] [CrossRef]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef]
- Li, G.; Wang, K.; Li, Y.; Ruan, J.; Wang, C.; Qian, Y.; Zu, S.; Dai, B.; Meng, Y.; Zhou, R.; et al. Role of eIF3a in 4-amino-2-trifluoromethyl-phenyl retinate-induced cell differentiation in human chronic myeloid leukemia K562 cells. Gene 2018, 683, 195–209. [Google Scholar] [CrossRef]
- Liu, T.; Wei, Q.; Jin, J.; Luo, Q.; Liu, Y.; Yang, Y.; Cheng, C.; Li, L.; Pi, J.; Si, Y.; et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020, 48, 3816–3831. [Google Scholar] [CrossRef]
- Qi, J.; Dong, Z.; Liu, J.; Zhang, J.-T. EIF3i promotes colon oncogenesis by regulating COX-2 protein synthesis and β-catenin activation. Oncogene 2013, 33, 4156–4163. [Google Scholar] [CrossRef][Green Version]
- Jiang, M.; Lu, Y.; Duan, D.; Wang, H.; Man, G.; Kang, C.; Abulimiti, K.; Li, Y. Systematic Investigation of mRNA N6-Methyladenosine Machinery in Primary Prostate Cancer. Dis. Mrk. 2020, 2020, 8833438. [Google Scholar] [CrossRef]
- Zhang, L.; Smit-McBride, Z.; Pan, X.; Rheinhardt, J.; Hershey, J.W.B. An Oncogenic Role for the Phosphorylated h-Subunit of Human Translation Initiation Factor eIF3. J. Biol. Chem. 2008, 283, 24047–24060. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Qiao, G.-L.; Zeng, X.-C.; Li, Y.; Yan, J.-J.; Duan, R.; Du, Z.-Y. Elevated expression of eukaryotic translation initiation factor 3H is associated with proliferation, invasion and tumorigenicity in human hepatocellular carcinoma. Oncotarget 2016, 7, 49888–49901. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef]
- Carvajal-Carmona, L.G.; Cazier, J.B.; Jones, A.M.; Howarth, K.; Broderick, P.; Pittman, A.; Dobbins, S.; Tenesa, A.; Farrington, S.; Prendergast, J.; et al. Fine-mapping of colorectal cancer susceptibility loci at 8q23.3, 16q22.1 and 19q13.11: Refinement of association signals and use of in silico analysis to suggest functional variation and unexpected candidate target genes. Hum. Mol. Genet. 2011, 20, 2879–2888. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Kordon, E.; Callahan, R.; Smith, G.H. Evidence for the transforming activity of a truncated Int6 gene, in vitro. Oncogene 2001, 20, 5291–5301. [Google Scholar] [CrossRef] [PubMed]
- Grzmil, M.; Rzymski, T.; Milani, M.; Harris, A.L.; Capper, R.G.; Saunders, N.J.; Salhan, A.; Ragoussis, J.; Norbury, C.J. An oncogenic role of eIF3e/INT6 in human breast cancer. Oncogene 2010, 29, 4080–4089. [Google Scholar] [CrossRef]
- Doldan, A.; Chandramouli, A.; Shanas, R.; Bhattacharyya, A.; Cunningham, J.T.; Nelson, M.A.; Shi, J. Loss of the eukaryotic initiation factor 3f in pancreatic cancer. Mol. Carcinog. 2008, 47, 235–244. [Google Scholar] [CrossRef]
- Shi, J.; Kahle, A.; Hershey, J.W.; Honchak, B.M.; Warneke, J.A.; Leong, S.P.; Nelson, M.A. Decreased expression of eukaryotic initiation factor 3f deregulates translation and apoptosis in tumor cells. Oncogene 2006, 25, 4923–4936. [Google Scholar] [CrossRef][Green Version]
- Sonenberg, N.; Dever, T.E. Eukaryotic translation initiation factors and regulators. Curr. Opin. Struct. Biol. 2003, 13, 56–63. [Google Scholar] [CrossRef]
- Rozovsky, N.; Butterworth, A.C.; Moore, M.J. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA 2008, 14, 2136–2148. [Google Scholar] [CrossRef]
- Chang, J.H.; Cho, Y.H.; Sohn, S.Y.; Choi, J.M.; Kim, A.; Kim, Y.C.; Jang, S.K.; Cho, Y. Crystal structure of the eIF4A–PDCD4 complex. Proc. Natl. Acad. Sci. USA 2009, 106, 3148–3153. [Google Scholar] [CrossRef] [PubMed]
- Harms, U.; Andreou, A.Z.; Gubaev, A.; Klostermeier, D. eIF4B, eIF4G and RNA regulate eIF4A activity in translation initiation by modulating the eIF4A conformational cycle. Nucleic Acids Res. 2014, 42, 7911–7922. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F Translation Initiation Complex: A Critical Nexus for Cancer Development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Demosthenous, C.; Han, J.J.; Stenson, M.J.; Maurer, M.J.; Wellik, L.E.; Link, B.; Hege, K.; Dogan, A.; Sotomayor, E.; Witzig, T.; et al. Translation initiation complex eIF4F is a therapeutic target for dual mTOR kinase inhibitors in non-Hodgkin lymphoma. Oncotarget 2015, 6, 9488–9501. [Google Scholar] [CrossRef]
- Shahbazian, D.; Parsyan, A.; Petroulakis, E.; Hershey, J.W.; Sonenberg, N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle 2010, 9, 4106–4109. [Google Scholar] [CrossRef]
- Kroczynska, B.; Kaur, S.; Katsoulidis, E.; Majchrzak-Kita, B.; Sassano, A.; Kozma, S.C.; Fish, E.N.; Platanias, L.C. Interferon-dependent engagement of eukaryotic initiation factor 4B via S6 kinase (S6K)- and ribosomal protein S6K-mediated signals. Mol. Cell. Biol. 2009, 29, 2865–2875. [Google Scholar] [CrossRef][Green Version]
- Wang, R.-T.; Xu, M.; Xu, C.-X.; Song, Z.-G.; Jin, H. Decreased Expression of miR216a Contributes to Non–Small-Cell Lung Cancer Progression. Clin. Cancer Res. 2014, 20, 4705–4716. [Google Scholar] [CrossRef]
- Shahbazian, D.; Parsyan, A.; Petroulakis, E.; Topisirovic, I.; Martineau, Y.; Gibbs, B.F.; Svitkin, Y.; Sonenberg, N. Control of Cell Survival and Proliferation by Mammalian Eukaryotic Initiation Factor 4B. Mol. Cell. Biol. 2010, 30, 1478–1485. [Google Scholar] [CrossRef]
- Ruggero, D.; Montanaro, L.; Ma, L.; Xu, W.; Londei, P.; Cordon-Cardo, C.; Pandolfi, P.P. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med. 2004, 10, 484–486. [Google Scholar] [CrossRef]
- Carroll, M.; Borden, K.L. The Oncogene eIF4E: Using Biochemical Insights to Target Cancer. J. Interf. Cytokine Res. 2013, 33, 227–238. [Google Scholar] [CrossRef]
- Bitterman, P.B.; Polunovsky, V.A. eIF4E-mediated translational control of cancer incidence. Biochim. Biophys. Acta 2015, 1849, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A Selective Inhibitor of eIF2α Dephosphorylation Protects Cells from ER Stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wolgamott, L.; Tcherkezian, J.; Vallabhapurapu, S.; Yu, Y.; Roux, P.P.; Yoon, S.-O. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 2013, 33, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.D.; Papadopoulos, E.; Dempersmier, J.M.; Wang, Z.-F.; Nowak, R.P.; Donovan, K.A.; Kalabathula, J.; Gorgulla, C.; Junghanns, P.P.; Kabha, E.; et al. A biphenyl inhibitor of eIF4E targeting an internal binding site enables the design of cell-permeable PROTAC-degraders. Eur. J. Med. Chem. 2021, 219, 113435. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Andrieu, C.; Ziouziou, H.; Rocchi, P. The Eukaryotic Translation Initiation Factor 4E (eIF4E) as a Therapeutic Target for Cancer. Adv. Protein Chem. Struct. Biol. 2015, 101, 1–26. [Google Scholar]
- Wang, H.; Liu, Y.; Ding, J.; Huang, Y.; Liu, J.; Liu, N.; Ao, Y.; Hong, Y.; Wang, L.; Zhang, L.; et al. Targeting mTOR suppressed colon cancer growth through 4EBP1/eIF4E/PUMA pathway. Cancer Gene Ther. 2020, 27, 448–460. [Google Scholar] [CrossRef]
- Guo, Q.; Bartish, M.; Gonçalves, C.; Huang, F.; Smith-Voudouris, J.; Krisna, S.S.; Preston, S.E.J.; Emond, A.; Li, V.Z.; Duerr, C.U.; et al. The MNK1/2-eIF4E Axis Supports Immune Suppres-sion and Metastasis in Postpartum Breast Cancer. Cancer Res. 2021, 81, 3876–3889. [Google Scholar] [CrossRef]
- Wan, J.; Shi, F.; Xu, Z.; Zhao, M. Knockdown of eIF4E suppresses cell proliferation, invasion and enhances cisplatin cytotoxicity in human ovarian cancer cells. Int. J. Oncol. 2015, 47, 2217–2225. [Google Scholar] [CrossRef]
- Graff, J.R.; Konicek, B.W.; Lynch, R.L.; Dumstorf, C.A.; Dowless, M.S.; McNulty, A.M.; Parsons, S.H.; Brail, L.H.; Colligan, B.M.; Koop, J.W.; et al. eIF4E Activation Is Commonly Elevated in Advanced Human Prostate Cancers and Significantly Related to Reduced Patient Survival. Cancer Res. 2009, 69, 3866–3873. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, S.; Chen, Z.; Wang, L.; Zhu, W.; Yin, C.; Fan, J.; Wu, X.; Wang, J.; Guo, C. EGPI-1, a novel eIF4E/eIF4G interaction inhibitor, inhibits lung cancer cell growth and angiogenesis through Ras/MNK/ERK/eIF4E signaling pathway. Chem. Interact. 2021, 352, 109773. [Google Scholar] [CrossRef] [PubMed]
- Herzog, L.-O.; Walters, B.; Buono, R.; Lee, J.S.; Mallya, S.; Fung, A.; Chiu, H.; Nguyen, N.; Li, B.; Pinkerton, A.B.; et al. Targeting eIF4F translation initiation complex with SBI-756 sensitises B lymphoma cells to venetoclax. Br. J. Cancer 2020, 124, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-I.; Wang, C.-C.; Tai, T.-S.; Hwang, T.-Z.; Yang, C.-C.; Hsu, C.-M.; Su, Y.-C. eIF4E and 4EBP1 are prognostic markers of head and neck squamous cell carcinoma recurrence after definitive surgery and adjuvant radiotherapy. PLoS ONE 2019, 14, e0225537. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chiu, T.-L.; Amin, E.A.; Polunovsky, V.; Bitterman, P.B.; Wagner, C.R. Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn7-GMP. Eur. J. Med. Chem. 2010, 45, 1304–1313. [Google Scholar] [CrossRef]
- Mao, C.; Liu, H.; Chen, P.; Ye, J.; Teng, L.; Jia, Z.; Cao, J. Cell-specific expression of artificial microRNAs targeting essential genes exhibit potent antitumor effect on hepatocellular carcinoma cells. Oncotarget 2015, 6, 5707–5719. [Google Scholar] [CrossRef]
- Gross, J.D.; Moerke, N.J.; von der Haar, T.; Lugovskoy, A.A.; Sachs, A.B.; McCarthy, J.E.; Wagner, G. Ribosome Loading onto the mRNA Cap Is Driven by Conformational Coupling between eIF4G and eIF4E. Cell 2003, 115, 739–750. [Google Scholar] [CrossRef]
- Wang, H.; Huang, F.; Wang, J.; Wang, P.; Lv, W.; Hong, L.; Li, S.; Zhou, J. The synergistic inhibition of breast cancer proliferation by combined treatment with 4EGI-1 and MK2206. Cell Cycle 2015, 14, 232–242. [Google Scholar] [CrossRef]
- Özeş, A.R.; Feoktistova, K.; Avanzino, B.C.; Fraser, C.S. Duplex Unwinding and ATPase Activities of the DEAD-Box Helicase eIF4A Are Coupled by eIF4G and eIF4B. J. Mol. Biol. 2011, 412, 674–687. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Tanaka, K.; Ryo, A.; Wakatsuki, T.; Hada, A.; Goseki, N.; Igari, T.; Hatsuse, K.; Aihara, T.; et al. Enhanced expression of translation factor mRNAs in hepatocellular carcinoma. Anticancer Res. 2000, 20, 2489–2494. [Google Scholar]
- Xue, C.; Gu, X.; Li, G.; Bao, Z.; Li, L. Expression and Functional Roles of Eukaryotic Initiation Factor 4A Family Proteins in Human Cancers. Front. Cell Dev. Biol. 2021, 9, 711965. [Google Scholar] [CrossRef]
- Conte, M.R.; Kelly, G.; Babon, J.; Sanfelice, D.; Youell, J.; Smerdon, S.J.; Proud, C.G. Structure of the Eukaryotic Initiation Factor (eIF) 5 Reveals a Fold Common to Several Translation Factors. Biochemistry 2006, 45, 4550–4558. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.D.; Pavitt, G.D. eIF5 has GDI activity necessary for translational control by eIF2 phosphorylation. Nature 2010, 465, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.X.; Chen, W.S.; Zeng, W.; Cheng, X.F.; Lin, M.B.; Wang, J.S. Roles of HDAC2, eIF5, and eIF6 in Lung Cancer Tumorigenesis. Curr. Med. Sci. 2021, 41, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Schuller, A.P.; Wu, C.C.-C.; Dever, T.E.; Buskirk, A.R.; Green, R. eIF5A Functions Globally in Translation Elongation and Termination. Mol. Cell 2017, 66, 194–205.e5. [Google Scholar] [CrossRef] [PubMed]
- Clement, P.M.J.; Henderson, C.A.; Jenkins, Z.A.; Smit-McBride, Z.; Wolff, E.C.; Hershey, J.W.B.; Park, M.H.; Johansson, H.E. Identification and characterization of eukaryotic initiation factor 5A-2. Eur. J. Biochem. 2003, 270, 4254–4263. [Google Scholar] [CrossRef]
- Clement, P.M.; Johansson, H.E.; Wolff, E.C.; Park, M.H. Differential expression of eIF5A-1 and eIF5A-2 in human cancer cells. FEBS J. 2006, 273, 1102–1114. [Google Scholar] [CrossRef]
- Bao, Y.; Lu, Y.; Wang, X.; Feng, W.; Sun, X.; Guo, H.; Tang, C.; Zhang, X.; Shi, Q.; Yu, H. Eukaryotic translation initiation fac-tor 5A2 (eIF5A2) regulates chemoresistance in colorectal cancer through epithelial mesenchymal transition. Cancer Cell Int. 2015, 15, 109. [Google Scholar] [CrossRef]
- Chukka, P.A.R.; Wetmore, S.D.; Thakor, N. Established and Emerging Regulatory Roles of Eukaryotic Translation Initiation Factor 5B (eIF5B). Front. Genet. 2021, 12, 737433. [Google Scholar] [CrossRef]
- Lee, S.; Truesdell, S.S.; Bukhari, S.I.A.; Lee, J.H.; LeTonqueze, O.; Vasudevan, S. Upregulation of eIF5B controls cell-cycle arrest and specific developmental stages. Proc. Natl. Acad. Sci. USA 2014, 111, E4315–E4322. [Google Scholar] [CrossRef]
- Ross, J.; Dungen, K.V.; Bressler, K.R.; Fredriksen, M.; Sharma, D.K.; Balasingam, N.; Thakor, N. Eukaryotic initiation factor 5B (eIF5B) provides a critical cell survival switch to glioblastoma cells via regulation of apoptosis. Cell Death Dis. 2019, 10, 57. [Google Scholar] [CrossRef]
- Suresh, S.; Chen, B.; Zhu, J.; Golden, R.J.; Lu, C.; Evers, B.M.; Novaresi, N.; Smith, B.; Zhan, X.; Schmid, V.; et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nat. Cancer 2020, 1, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-G.; Zheng, H.; Gao, W.; Han, J.; Cao, J.-Z.; Yang, Y.; Li, S.; Gao, R.; Liu, H.; Pan, Z.-Y.; et al. eIF5B increases ASAP1 expression to promote HCC proliferation and invasion. Oncotarget 2016, 7, 62327–62339. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Wu, J.; Liu, J.; Qin, Z.; Fan, H. N6-Methyladenosine: A Potential Breakthrough for Human Cancer. Mol. Ther. Nucleic Acids 2019, 19, 804–813. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 2018, 22, 191–205.e199. [Google Scholar] [CrossRef] [PubMed]
- Bujnicki, J.M.; Feder, M.; Radlinska, M.; Blumenthal, R.M. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol. 2002, 55, 431–444. [Google Scholar] [CrossRef]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Corrigendum: Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 2017, 542, 260. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Li, J.; Chen, R.; Gu, Q.; Yang, P.; Qian, W.; Ji, D.; Wang, Q.; Zhang, Z.; Tang, J.; et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 393. [Google Scholar] [CrossRef]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell. Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef]
- Li, F.; Yi, Y.; Miao, Y.; Long, W.; Long, T.; Chen, S.; Cheng, W.; Zou, C.; Zheng, Y.; Wu, X.; et al. N(6)-Methyladenosine Modulates Nonsense-Mediated mRNA Decay in Human Glioblastoma. Cancer Res. 2019, 79, 5785–5798. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hu, P.S.; Zuo, Z.; Lin, J.F.; Li, X.; Wu, Q.N.; Chen, Z.H.; Zeng, Z.L.; Wang, F.; Zheng, J.; et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer 2019, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Luo, G.; Xu, W.; Zhao, Y.; Jin, S.; Wang, S.; Liu, Q.; Chen, X.; Wang, J.; Dong, F.; Hu, D.N.; et al. RNA m(6) A methylation regulates uveal melanoma cell proliferation, migration, and invasion by targeting c-Met. J. Cell. Physiol. 2020, 235, 7107–7119. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Chen, J.; Jia, L.; Ma, J.; Song, D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem. Biophys. Res. Commun. 2019, 516, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Cai, J.; Yang, F.; Zhan, H.; Situ, J.; Li, W.; Mao, Y.; Luo, Y. RNA m(6)A Methyltransferase METTL3 Promotes The Growth Of Prostate Cancer By Regulating Hedgehog Pathway. Onco Targets 2019, 12, 9143–9152. [Google Scholar] [CrossRef]
- Li, E.; Wei, B.; Wang, X.; Kang, R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am. J. Cancer Res. 2020, 10, 1012–1025. [Google Scholar]
- Wang, J.; Zhang, C.; He, W.; Gou, X. Effect of m(6)A RNA Methylation Regulators on Malignant Progression and Prognosis in Renal Clear Cell Carcinoma. Front. Oncol. 2020, 10, 3. [Google Scholar] [CrossRef]
- Li, X.; Tang, J.; Huang, W.; Wang, F.; Li, P.; Qin, C.; Qin, Z.; Zou, Q.; Wei, J.; Hua, L.; et al. The M6A methyltransferase METTL3: Acting as a tumor suppressor in renal cell carcinoma. Oncotarget 2017, 8, 96103–96116. [Google Scholar] [CrossRef]
- Visvanathan, A.; Patil, V.; Arora, A.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 2018, 37, 522–533. [Google Scholar] [CrossRef]
- Gu, C.; Wang, Z.; Zhou, N.; Li, G.; Kou, Y.; Luo, Y.; Wang, Y.; Yang, J.; Tian, F. Mettl14 inhibits bladder TIC self-renewal and bladder tumorigenesis through N(6)-methyladenosine of Notch1. Mol. Cancer 2019, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, M.; Xu, X.; Zeng, K.; Liu, X.; Sun, L.; Pan, B.; He, B.; Pan, Y.; Sun, H.; et al. RETRACTED: METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol. Ther. 2020, 28, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, S.; He, C.; Xue, P.; Zhang, L.; He, Z.; Zang, L.; Feng, B.; Sun, J.; Zheng, M. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer 2020, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Zhang, J.; Chen, Y.; Xu, Y.; Ma, J.; Hu, G.; Huang, Y.; Zheng, J.; Zhai, W.; Xue, W. The m(6)A-suppressed P2RX6 activation promotes renal cancer cells migration and invasion through ATP-induced Ca(2+) influx modulating ERK1/2 phosphorylation and MMP9 signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 233. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, J.; Zhao, Y.; He, R.; Xu, X.; Guo, X.; Li, X.; Xu, S.; Miao, J.; Guo, J.; et al. Upregulation of METTL14 mediates the elevation of PERP mRNA N(6) adenosine methylation promoting the growth and metastasis of pancreatic cancer. Mol. Cancer 2020, 19, 130. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Tao, L.; Mu, X.; Chen, H.; Jin, D.; Zhang, R.; Zhao, Y.; Fan, J.; Cao, M.; Zhou, Z. FTO modifies the m6A level of MALAT and promotes bladder cancer progression. Clin. Transl. Med. 2021, 11, e310. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.F.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef]
- Zou, D.; Dong, L.; Li, C.; Yin, Z.; Rao, S.; Zhou, Q. The m(6)A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019, 19, 321. [Google Scholar] [CrossRef]
- Bian, X.; Shi, D.; Xing, K.; Zhou, H.; Lu, L.; Yu, D.; Wu, W. AMD1 upregulates hepatocellular carcinoma cells stemness by FTO mediated mRNA demethylation. Clin. Transl. Med. 2021, 11, e352. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ren, D.; Du, Z.; Wang, H.; Zhang, H.; Jin, Y. m(6)A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem. Biophys. Res. Commun. 2018, 502, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, J.; Cui, Y.H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef]
- Tang, X.; Liu, S.; Chen, D.; Zhao, Z.; Zhou, J. The role of the fat mass and obesity-associated protein in the proliferation of pancreatic cancer cells. Oncol. Lett. 2019, 17, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Sheng, Y.; Zhu, A.C.; Robinson, S.; Jiang, X.; Dong, L.; Chen, H.; Su, R.; Yin, Z.; Li, W.; et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020, 27, 64–80.e69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e596. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, S.; Piao, H.Y.; Wang, Y.; Wu, Y.; Meng, X.Y.; Yang, D.; Zheng, Z.C.; Zhao, Y. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J. Physiol. Biochem. 2019, 75, 379–389. [Google Scholar] [CrossRef]
- Jin, D.; Guo, J.; Wu, Y.; Yang, L.; Wang, X.; Du, J.; Dai, J.; Chen, W.; Gong, K.; Miao, S.; et al. m(6)A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol. Cancer 2020, 19, 40. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, L.; Wang, Y. ALKBH5-mediated m(6)A demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020, 20, 34. [Google Scholar] [CrossRef]
- Jiang, Y.; Wan, Y.; Gong, M.; Zhou, S.; Qiu, J.; Cheng, W. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-κB pathway. J. Cell. Mol. Med. 2020, 24, 6137–6148. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Yang, Y.; Kang, M.; Wang, Y.; Wang, Y.; Bi, Y.; He, S.; Shimamoto, F. m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol. Cancer 2020, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Tanikawa, K.; Tsunetomi, M.; Takai, K.; Ikeda, H.; Konno, J.; Torigoe, T.; Maeda, H.; Kutomi, G.; Okita, K.; et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. 2016, 376, 34–42. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, C.; Wu, R.; Huang, L.; Song, S.; Li, W.; Yan, P.; Lin, C.; Li, D.; Zhang, Y. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front. Oncol. 2019, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Chai, P.; Wang, S.; Sun, B.; Xu, Y.; Yang, Y.; Ge, S.; Jia, R.; Yang, Y.G.; Fan, X. m(6)A modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol. Cancer 2019, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148.e136. [Google Scholar] [CrossRef]
- Zhong, L.; Liao, D.; Zhang, M.; Zeng, C.; Li, X.; Zhang, R.; Ma, H.; Kang, T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019, 442, 252–261. [Google Scholar] [CrossRef]
- Sheng, H.; Li, Z.; Su, S.; Sun, W.; Zhang, X.; Li, L.; Li, J.; Liu, S.; Lu, B.; Zhang, S.; et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis 2020, 41, 541–550. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Cai, X.; Wang, X.; Cao, C.; Gao, Y.; Zhang, S.; Yang, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Ye, L. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018, 415, 11–19. [Google Scholar] [CrossRef]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.T.; Xu, Q.G.; Zhou, W.P.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, X.; Chen, Z.; Tian, L.; Jiang, G.; Chen, F.; Li, J.; An, P.; Lu, L.; Luo, N.; et al. m6A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol. Cancer 2019, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, L.; Dong, Z.; Li, J.; Yu, Y.; Chen, X.; Ren, F.; Cui, G.; Sun, R. Expression patterns and prognostic value of m6A-related genes in colorectal cancer. Am. J. Transl. Res. 2019, 11, 3972–3991. [Google Scholar]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 14013–14018. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.; Lynch, K.W. Alternative splicing and cancer: Insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020, 34, 1005–1016. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, L.J.; Qiang, P. The Potential Role of N6-Methyladenosine (m6A) Demethylase Fat Mass and Obesity-Associated Gene (FTO) in Human Cancers. Onco Targets Ther. 2020, 13, 12845–12856. [Google Scholar] [CrossRef]
- Zhang, C.; Zhi, W.I.; Lu, H.; Samanta, D.; Chen, I.; Gabrielson, E.; Semenza, G.L. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 2016, 7, 64527–64542. [Google Scholar] [CrossRef]
- Zhu, H.; Gan, X.; Jiang, X.; Diao, S.; Wu, H.; Hu, J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 2019, 38, 163. [Google Scholar] [CrossRef]
- Wei, C.-M.; Gershowitz, A.; Moss, B. N6, O2′-dimethyladenosine a novel methylated ribonucleoside next to the 5′ terminal of animal cell and virus mRNAs. Nature 1975, 257, 251–253. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, M.; Li, K.; Bai, D.; Yi, C. Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res. 2019, 29, 80–82. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.-J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Sindelar, M.; Despic, V.; Guez, T.; Hawley, B.R.; Vasseur, J.-J.; Rentmeister, A.; Gross, S.S.; Pellizzoni, L.; Debart, F.; et al. FTO controls reversible m6Am RNA methylation during snRNA biogenesis. Nat. Chem. Biol. 2019, 15, 340–347. [Google Scholar] [CrossRef]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef] [PubMed]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Nureki, O.; et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 2019, 363, eaav0080. [Google Scholar] [CrossRef] [PubMed]
- Relier, S.; Ripoll, J.; Guillorit, H.; Amalric, A.; Achour, C.; Boissière, F.; Vialaret, J.; Attina, A.; Debart, F.; Choquet, A.; et al. FTO-mediated cytoplasmic m(6)A(m) demethylation adjusts stem-like properties in colorectal cancer cell. Nat. Commun. 2021, 12, 1716. [Google Scholar] [CrossRef] [PubMed]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.J.; Debart, F.; Aravind, L.; et al. Identification of the m(6)Am Methyltransferase PCIF1 Reveals the Location and Functions of m(6)Am in the Transcriptome. Mol. Cell 2019, 75, 631–643.e638. [Google Scholar] [CrossRef]
- Gao, S.; Zhou, J.; Hu, Z.; Zhang, S.; Wu, Y.; Musunuru, P.P.; Zhang, T.; Yang, L.; Luo, X.; Bai, J.; et al. Effects of the m6Am methyltransferase PCIF1 on cell proliferation and survival in gliomas. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166498. [Google Scholar] [CrossRef]
- Zhuo, W.; Sun, M.; Wang, K.; Zhang, L.; Li, K.; Yi, D.; Li, M.; Sun, Q.; Ma, X.; Liu, W.; et al. m(6)Am methyltransferase PCIF1 is essential for aggressiveness of gastric cancer cells by inhibiting TM9SF1 mRNA translation. Cell Discov. 2022, 8, 48. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Zhang, Y.-G.; Jin, W.-L.; Wang, X.-P. A Pan-Cancer Analysis of the Oncogenic and Immunogenic Role of m6Am Methyltransferase PCIF1. Front. Oncol. 2021, 11, 753393. [Google Scholar] [CrossRef]
- García-Vílchez, R.; Sevilla, A.; Blanco, S. Post-transcriptional regulation by cytosine-5 methylation of RNA. Biochim. Biophys. Acta 2018, 1862, 240–252. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m⁵C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Dragoni, I.; Chin, S.F.; Spiteri, I.; Kurowski, A.; Provenzano, E.; Green, A.; Ellis, I.O.; Grimmer, D.; Teschendorff, A.; et al. Genomic gain of 5p15 leads to over-expression of Misu (NSUN2) in breast cancer. Cancer Lett. 2010, 289, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Sato, G.; Usui, K.; Sato, M.; Sagawa, M.; Kondo, T.; Minami, Y.; Nukiwa, T. Expression of nucleolar protein p120 predicts poor prognosis in patients with stage I lung adenocarcinoma. Ann. Oncol. 2001, 12, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Bantis, A.; Giannopoulos, A.; Gonidi, M.; Liossi, A.; Aggelonidou, E.; Petrakakou, E.; Athanassiades, P.; Athanassiadou, P. Expression of p120, Ki-67 and PCNA as proliferation biomarkers in imprint smears of prostate carcinoma and their prognostic value. Cytopathology 2004, 15, 25–31. [Google Scholar] [CrossRef]
- Cheng, J.X.; Chen, L.; Li, Y.; Cloe, A.; Yue, M.; Wei, J.; Watanabe, K.A.; Shammo, J.M.; Anastasi, J.; Shen, Q.J.; et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018, 9, 1163. [Google Scholar] [CrossRef]
- Chen, X.; Li, A.; Sun, B.F.; Yang, Y.; Han, Y.N.; Yuan, X.; Chen, R.X.; Wei, W.S.; Liu, Y.; Gao, C.C.; et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 2019, 21, 978–990. [Google Scholar] [CrossRef]
- Frye, M.; Watt, F.M. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. 2006, 16, 971–981. [Google Scholar] [CrossRef]
- Blanco, S.; Bandiera, R.; Popis, M.; Hussain, S.; Lombard, P.; Aleksic, J.; Sajini, A.; Tanna, H.; Cortés-Garrido, R.; Gkatza, N.; et al. Stem cell function and stress response are controlled by protein synthesis. Nature 2016, 534, 335–340. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Z.; Zhu, Y.; Zhu, Q.; Yang, Y.; Jin, Y.; Zhang, F.; Jiang, L.; Ye, Y.; Li, H.; et al. NOP2/Sun RNA methyltransferase 2 promotes tumor progression via its interacting partner RPL6 in gallbladder carcinoma. Cancer Sci. 2019, 110, 3510–3519. [Google Scholar] [CrossRef]
- Mei, L.; Shen, C.; Miao, R.; Wang, J.Z.; Cao, M.D.; Zhang, Y.S.; Shi, L.H.; Zhao, G.H.; Wang, M.H.; Wu, L.S.; et al. RNA methyltransferase NSUN2 promotes gastric cancer cell proliferation by repressing p57(Kip2) by an m(5)C-dependent manner. Cell Death Dis. 2020, 11, 270. [Google Scholar] [CrossRef]
- Lu, L.; Zhu, G.; Zeng, H.; Xu, Q.; Holzmann, K. High tRNA Transferase NSUN2 Gene Expression is Associated with Poor Prognosis in Head and Neck Squamous Carcinoma. Cancer Investig. 2018, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Luo, M.; Zhou, C.; Shi, X.; Yang, W.; Lu, Z.; Chen, Z.; Sun, N.; He, J. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 2018, 430, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Risch, E.; Zhang, M.; Huang, C.; Huang, H.; Lu, L. Association of tRNA methyltransferase NSUN2/IGF-II molecular signature with ovarian cancer survival. Future Oncol. 2017, 13, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hirata, S.; Sato, S.; Koga, S.; Fujii, M.; Qi, G.; Ogawa, I.; Takata, T.; Shimamoto, F.; Tatsuka, M. Frequent increased gene copy number and high protein expression of tRNA (cytosine-5-)-methyltransferase (NSUN2) in human cancers. DNA Cell Biol. 2012, 31, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, X.; Li, J.; Zhang, Q.; Zheng, Q.; Guo, W. Role of m(5)C-related regulatory genes in the diagnosis and prognosis of hepatocellular carcinoma. Am. J. Transl. Res. 2020, 12, 912–922. [Google Scholar]
- Sato, K.; Tahata, K.; Akimoto, K. Five Genes Associated With Survival in Patients With Lower-grade Gliomas Were Identified by Information-theoretical Analysis. Anticancer Res. 2020, 40, 2777–2785. [Google Scholar] [CrossRef]
- Elhardt, W.; Shanmugam, R.; Jurkowski, T.P.; Jeltsch, A. Somatic cancer mutations in the DNMT2 tRNA methyltransferase alter its catalytic properties. Biochimie 2015, 112, 66–72. [Google Scholar] [CrossRef]
- Wang, C.; Huang, Y.; Zhang, J.; Fang, Y. MiRNA-339-5p suppresses the malignant development of gastric cancer via targeting ALKBH1. Exp. Mol. Pathol. 2020, 115, 104449. [Google Scholar] [CrossRef]
- García, M.G.; Carella, A.; Urdinguio, R.G.; Bayón, G.F.; Lopez, V.; Tejedor, J.R.; Sierra, M.I.; García-Toraño, E.; Santamarina, P.; Perez, R.F.; et al. Epigenetic dysregulation of TET2 in human glioblastoma. Oncotarget 2018, 9, 25922–25934. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Carella, A.; Tejedor, J.R.; García, M.G.; Urdinguio, R.G.; Bayón, G.F.; Sierra, M.; López, V.; García-Toraño, E.; Santamarina-Ojeda, P.; Pérez, R.F.; et al. Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. Int. J. Cancer 2020, 146, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, D.; Wang, F.; Xu, Y.; Yu, H.; Zhang, H. Expression of Demethylase Genes, FTO and ALKBH1, Is Associated with Prognosis of Gastric Cancer. Dig. Dis. Sci. 2019, 64, 1503–1513. [Google Scholar] [CrossRef]
- Yamashita, T.; Higashi, M.; Momose, S.; Morozumi, M.; Tamaru, J.I. Nuclear expression of Y box binding-1 is important for resistance to chemotherapy including gemcitabine in TP53-mutated bladder cancer. Int. J. Oncol. 2017, 51, 579–586. [Google Scholar] [CrossRef]
- Campbell, T.M.; Castro, M.A.A.; de Oliveira, K.G.; Ponder, B.A.J.; Meyer, K.B. ERα Binding by Transcription Factors NFIB and YBX1 Enables FGFR2 Signaling to Modulate Estrogen Responsiveness in Breast Cancer. Cancer Res. 2018, 78, 410–421. [Google Scholar] [CrossRef]
- Kuwano, M.; Shibata, T.; Watari, K.; Ono, M. Oncogenic Y-box binding protein-1 as an effective therapeutic target in drug-resistant cancer. Cancer Sci. 2019, 110, 1536–1543. [Google Scholar] [CrossRef]
- Saito, Y.; Kasamatsu, A.; Yamamoto, A.; Shimizu, T.; Yokoe, H.; Sakamoto, Y.; Ogawara, K.; Shiiba, M.; Tanzawa, H.; Uzawa, K. ALY as a potential contributor to metastasis in human oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Sánchez, M.S.; Sáez, C.; Japón, M.A.; Aguilera, A.; Luna, R. Differential expression of THOC1 and ALY mRNP biogenesis/export factors in human cancers. BMC Cancer 2011, 11, 77. [Google Scholar] [CrossRef]
- Perlaky, L.; Valdez, B.C.; Busch, R.K.; Larson, R.G.; Jhiang, S.M.; Zhang, W.W.; Brattain, M.; Busch, H. Increased growth of NIH/3T3 cells by transfection with human p120 complementary DNA and inhibition by a p120 antisense construct. Cancer Res. 1992, 52, 428–436. [Google Scholar]
- Freeman, J.W.; McGrath, P.; Bondada, V.; Selliah, N.; Ownby, H.; Maloney, T.; Busch, R.K.; Busch, H. Prognostic significance of proliferation associated nucleolar antigen P120 in human breast carcinoma. Cancer Res. 1991, 51, 1973–1978. [Google Scholar]
- Hong, J.; Lee, J.H.; Chung, I.K. Telomerase activates transcription of cyclin D1 gene through the interaction with NOL1. J. Cell Sci. 2016, 129, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. mRNA methylation by NSUN2 in cell proliferation. Wiley Interdiscip. Rev. RNA 2016, 7, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Janin, M.; Ortiz-Barahona, V.; de Moura, M.C.; Martínez-Cardús, A.; Llinàs-Arias, P.; Soler, M.; Nachmani, D.; Pelletier, J.; Schumann, U.; Calleja-Cervantes, M.E.; et al. Epigenetic loss of RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a stress adaptive translational program. Acta Neuropathol. 2019, 138, 1053–1074. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.V.; Cordero-Espinoza, L.; Oeztuerk-Winder, F.; Andersson-Rolf, A.; Selmi, T.; Blanco, S.; Tailor, J.; Dietmann, S.; Frye, M. Cytosine-5 RNA Methylation Regulates Neural Stem Cell Differentiation and Motility. Stem Cell Rep. 2016, 8, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Rosace, D.; López, J.; Blanco, S. Emerging roles of novel small non-coding regulatory RNAs in immunity and cancer. RNA Biol. 2020, 17, 1196–1213. [Google Scholar] [CrossRef]
- Lyons, S.M.; Gudanis, D.; Coyne, S.M.; Gdaniec, Z.; Ivanov, P. Identification of functional tetramolecular RNA G-quadruplexes derived from transfer RNAs. Nat. Commun. 2017, 8, 1127. [Google Scholar] [CrossRef]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-Induced tRNA Fragments Inhibit Translation Initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef]
- Hussain, S.; Tuorto, F.; Menon, S.; Blanco, S.; Cox, C.; Flores, J.V.; Watt, S.; Kudo, N.R.; Lyko, F.; Frye, M. The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol. Cell. Biol. 2013, 33, 1561–1570. [Google Scholar] [CrossRef]
- Ramon, Y.C.S.; Castellvi, J.; Hümmer, S.; Peg, V.; Pelletier, J.; Sonenberg, N. Beyond molecular tumor heterogeneity: Protein synthesis takes control. Oncogene 2018, 37, 2490–2501. [Google Scholar] [CrossRef]
- Archer, N.P.; Perez-Andreu, V.; Scheurer, M.E.; Rabin, K.R.; Peckham-Gregory, E.C.; Plon, S.E.; Zabriskie, R.C.; De Alarcon, P.A.; Fernandez, K.S.; Najera, C.R.; et al. Family-based exome-wide assessment of maternal genetic effects on susceptibility to childhood B-cell acute lymphoblastic leukemia in hispanics. Cancer 2016, 122, 3697–3704. [Google Scholar] [CrossRef]
- Li, W.; Xu, L. Epigenetic Function of TET Family, 5-Methylcytosine, and 5-Hydroxymethylcytosine in Hematologic Malignancies. Oncol. Res. Treat. 2019, 42, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Masuda, K.; Sato, T.; Sakaguchi, Y.; Suzuki, T.; Suzuki, T.; Koyama-Nasu, R.; Nasu-Nishimura, Y.; Katou, Y.; Ogawa, H.; et al. 5-Hydroxymethylcytosine Plays a Critical Role in Glioblastomagenesis by Recruiting the CHTOP-Methylosome Complex. Cell Rep. 2014, 9, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hamma, T.; Ferré-D’Amaré, A.R. Pseudouridine synthases. Chem. Biol. 2006, 13, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.F.; Allen, F.W. Ribonucleic Acids From Yeast Which Contain a Fifth Nucleotide. J. Biol. Chem. 1957, 227, 907–915. [Google Scholar] [CrossRef]
- Zhao, B.S.; He, C. Pseudouridine in a new era of RNA modifications. Cell Res. 2014, 25, 153–154. [Google Scholar] [CrossRef]
- Spenkuch, F.; Motorin, Y.; Helm, M. Pseudouridine: Still mysterious, but never a fake (uridine)! RNA Biol. 2014, 11, 1540–1554. [Google Scholar] [CrossRef]
- Hur, S.; Stroud, R.M.; Finer-Moore, J. Substrate recognition by RNA 5-methyluridine methyltransferases and pseudouridine synthases: A structural perspective. J. Biol. Chem. 2006, 281, 38969–38973. [Google Scholar] [CrossRef]
- Rintala-Dempsey, A.C.; Kothe, U. Eukaryotic stand-alone pseudouridine synthases-RNA modifying enzymes and emerging regulators of gene expression? RNA Biol. 2017, 14, 1185–1196. [Google Scholar] [CrossRef]
- Rostami, P.; Zendehdel, K.; Shirkoohi, R.; Ebrahimi, E.; Ataei, M.; Imanian, H.; Najmabadi, H.; Akbari, M.R.; Sanati, M.H. Gene Panel Testing in Hereditary Breast Cancer. Arch Iran Med. 2020, 23, 155–162. [Google Scholar]
- Poncet, D.; Belleville, A.; t’kint de Roodenbeke, C.; Roborel de Climens, A.; Ben Simon, E.; Merle-Beral, H.; Callet-Bauchu, E.; Salles, G.; Sabatier, L.; Delic, J.; et al. Changes in the expression of telomere maintenance genes suggest global telomere dysfunction in B-chronic lymphocytic leukemia. Blood 2008, 111, 2388–2391. [Google Scholar] [CrossRef]
- Kan, G.; Wang, Z.; Sheng, C.; Yao, C.; Mao, Y.; Chen, S. Inhibition of DKC1 induces telomere-related senescence and apoptosis in lung adenocarcinoma. J. Transl. Med. 2021, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Nersisyan, L.; Hopp, L.; Loeffler-Wirth, H.; Galle, J.; Loeffler, M.; Arakelyan, A.; Binder, H. Telomere Length Maintenance and Its Transcriptional Regulation in Lynch Syndrome and Sporadic Colorectal Carcinoma. Front. Oncol. 2019, 9, 1172. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.A.; Chu, K.; Chen, H.R.; Zhang, M.; Shi, P.C.; Bai, J.; You, Y.P. Increased DKC1 expression in glioma and its significance in tumor cell proliferation, migration and invasion. Investig. New Drugs 2019, 37, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Alawi, F.; Lin, P.; Ziober, B.; Patel, R. Correlation of dyskerin expression with active proliferation independent of telomerase. Head Neck 2011, 33, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, J.; Huang, C.; Liu, H. Dyskerin overexpression in human hepatocellular carcinoma is associated with advanced clinical stage and poor patient prognosis. PLoS ONE 2012, 7, e43147. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Keel, S.B.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Watts, A.C.; Pritchard, C.C.; Salipante, S.J.; Jeng, M.R.; Hofmann, I.; et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica 2015, 100, 42–48. [Google Scholar] [CrossRef]
- Stockert, J.A.; Gupta, A.; Herzog, B.; Yadav, S.S.; Tewari, A.K.; Yadav, K.K. Predictive value of pseudouridine in prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 262–272. [Google Scholar]
- Bellodi, C.; Krasnykh, O.; Haynes, N.; Theodoropoulou, M.; Peng, G.; Montanaro, L.; Ruggero, D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 2010, 70, 6026–6035. [Google Scholar] [CrossRef]
- Zhao, X.; Patton, J.R.; Davis, S.L.; Florence, B.; Ames, S.J.; Spanjaard, R.A. Regulation of nuclear receptor activity by a pseudouridine synthase through posttranscriptional modification of steroid receptor RNA activator. Mol. Cell 2004, 15, 549–558. [Google Scholar] [CrossRef]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef]
- Guzzi, N.; Cieśla, M.; Ngoc, P.C.T.; Lang, S.; Arora, S.; Dimitriou, M.; Pimková, K.; Sommarin, M.N.E.; Munita, R.; Lubas, M.; et al. Pseudouridylation of tRNA-Derived Fragments Steers Translational Control in Stem Cells. Cell 2018, 173, 1204–1216.e1226. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Hsieh, A.C.; Gupta, R. Reciprocal amplification of caspase-3 activity by nuclear export of a putative human RNA-modifying protein, PUS10 during TRAIL-induced apoptosis. Cell Death Dis. 2017, 8, e3093. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Ding, D.; Qin, N.; Wang, C.; Zhu, M.; Li, Y.; Dai, J.; Jin, G.; Hu, Z.; Shen, H.; et al. Systematic analyses of genetic variants in chromatin interaction regions identified four novel lung cancer susceptibility loci. J. Cancer 2020, 11, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Casoli, L.; Ceccarelli, C.; Treré, D.; Ludovini, V.; Crinò, L.; Montanaro, L. DKC1 gene mutations in human sporadic cancer. Histol. Histopathol. 2013, 28, 365–372. [Google Scholar]
- Dokal, I. Dyskeratosis congenita in all its forms. Br. J. Haematol. 2000, 110, 768–779. [Google Scholar] [CrossRef]
- Bellodi, C.; Kopmar, N.; Ruggero, D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010, 29, 1865–1876. [Google Scholar] [CrossRef]
- Rocchi, L.; Pacilli, A.; Sethi, R.; Penzo, M.; Schneider, R.J.; Treré, D.; Brigotti, M.; Montanaro, L. Dyskerin depletion increases VEGF mRNA internal ribosome entry site-mediated translation. Nucleic Acids Res. 2013, 41, 8308–8318. [Google Scholar] [CrossRef]
- McMahon, M.; Contreras, A.; Holm, M.; Uechi, T.; Forester, C.M.; Pang, X.; Jackson, C.; Calvert, M.E.; Chen, B.; Quigley, D.A.; et al. A single H/ACA small nucleolar RNA mediates tumor suppression downstream of oncogenic RAS. eLife 2019, 8, e48847. [Google Scholar] [CrossRef]
- Babaian, A.; Rothe, K.; Girodat, D.; Minia, I.; Djondovic, S.; Milek, M.; Miko, S.E.S.; Wieden, H.-J.; Landthaler, M.; Morin, G.B.; et al. Loss of m1acp3Ψ Ribosomal RNA Modification Is a Major Feature of Cancer. Cell Rep. 2020, 31, 107611. [Google Scholar] [CrossRef]
- Montanaro, L.; Brigotti, M.; Clohessy, J.; Barbieri, S.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Calienni, M.; Teruya-Feldstein, J.; Trerè, D.; et al. Dyskerin expression influences the level of ribosomal RNA pseudo-uridylation and telomerase RNA component in human breast cancer. J. Pathol. 2006, 210, 10–18. [Google Scholar] [CrossRef]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 11, a032896. [Google Scholar] [CrossRef] [PubMed]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886.e24. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, M.; Zhang, Y.; Xie, Y.; Zou, J.; Zhong, J.; Zheng, Z.; Zhou, X.; Zheng, Y.; Chen, B.; et al. NAT10-mediated mRNA N4-acetylcytidine modification promotes bladder cancer progression. Clin. Transl. Med. 2022, 12, e738. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zheng, M.H.; Zheng, Y.F.; Lu, A.G.; Li, J.W.; Wang, M.L.; Ma, J.J.; Xu, G.W.; Yu, B.M. Application of urinary nucleosides in the diagnosis and surgical monitoring of colorectal cancer. Zhonghua Wai Ke Za Zhi 2005, 43, 564–568. [Google Scholar]
- Liu, S.; Zhang, Y.; Qiu, L.; Zhang, S.; Meng, Y.; Huang, C.; Chen, Z.; Zhang, B.; Han, J. Uncovering N4-Acetylcytidine-Related mRNA Modification Pattern and Landscape of Stemness and Immunity in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2022, 10, 861000. [Google Scholar] [CrossRef]
- Zhang, Y.; Jing, Y.; Wang, Y.; Tang, J.; Zhu, X.; Jin, W.L.; Wang, Y.; Yuan, W.; Li, X.; Li, X. NAT10 promotes gastric cancer metastasis via N4-acetylated COL5A1. Signal Transduct. Target. Ther. 2021, 6, 173. [Google Scholar] [CrossRef]
- Yang, C.; Wu, T.; Zhang, J.; Liu, J.; Zhao, K.; Sun, W.; Zhou, X.; Kong, X.; Shi, J. Prognostic and Immunological Role of mRNA ac4C Regulator NAT10 in Pan-Cancer: New Territory for Cancer Research? Front. Oncol. 2021, 11, 630417. [Google Scholar] [CrossRef]
- Cho, S.; Lee, G.; Pickering, B.F.; Jang, C.; Park, J.H.; He, L.; Mathur, L.; Kim, S.-S.; Jung, S.; Tang, H.-W.; et al. mTORC1 promotes cell growth via m6A-dependent mRNA degradation. Mol. Cell 2021, 81, 2064–2075.e8. [Google Scholar] [CrossRef]
- Chen, H.; Gao, S.; Liu, W.; Wong, C.C.; Wu, J.; Wu, J.; Liu, D.; Gou, H.; Kang, W.; Zhai, J.; et al. RNA N(6)-Methyladenosine Methyltransferase METTL3 Facilitates Colorectal Cancer by Activating the m(6)A-GLUT1-mTORC1 Axis and Is a Therapeutic Target. Gastroenterology 2021, 160, 1284–1300.e1216. [Google Scholar] [CrossRef]
- Li, X.; Li, N.; Huang, L.; Xu, S.; Zheng, X.; Hamsath, A.; Zhang, M.; Dai, L.; Zhang, H.; Wong, J.J.; et al. Is Hydrogen Sulfide a Concern During Treatment of Lung Adenocarcinoma With Ammonium Tetrathiomolybdate? Front. Oncol. 2020, 10, 234. [Google Scholar] [CrossRef]
- Wei, W.; Sun, J.; Zhang, H.; Xiao, X.; Huang, C.; Wang, L.; Zhong, H.; Jiang, Y.; Zhang, X.; Jiang, G. Circ0008399 Interaction with WTAP Promotes Assembly and Activity of the m(6)A Methyltransferase Complex and Promotes Cisplatin Resistance in Bladder Cancer. Cancer Res. 2021, 81, 6142–6156. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhu, Y.; Cai, H.; Liang, J.; Wang, W.; Liao, Y.; Zhang, Y.; Wang, C.; Hou, J. N6-Methyladenosine Methyltransferase METTL14-Mediated Autophagy in Malignant Development of Oral Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 738406. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; He, P.C.; Liu, W.; Wei, J.; Zhao, Z.; Gao, L.; Han, L.; et al. METTL16 exerts an m(6)A-independent function to facilitate translation and tumorigenesis. Nat. Cell Biol. 2022, 24, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.-L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Orouji, E.; Peitsch, W.K.; Orouji, A.; Houben, R.; Utikal, J. Oncogenic Role of an Epigenetic Reader of m(6)A RNA Modification: YTHDF1 in Merkel Cell Carcinoma. Cancers 2020, 12, 202. [Google Scholar] [CrossRef]
- Kuai, D.; Zhu, S.; Shi, H.; Yang, R.; Liu, T.; Liu, H.; Min, L.; Zhang, S. Aberrant expression of m6A mRNA methylation regulators in colorectal adenoma and adenocarcinoma. Life Sci. 2021, 273, 119258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, H.; Zhang, D.; Quan, Q.; Ge, Y.; Li, L.; Guo, L. YTHDF3 Facilitates eIF2AK2 and eIF3A Recruitment on mRNAs to Regulate Translational Processes in Oxaliplatin-Resistant Colorectal Cancer. ACS Chem. Biol. 2022, 17, 1778–1788. [Google Scholar] [CrossRef]
- Wang, F.; Liao, Y.; Zhang, M.; Zhu, Y.; Wang, W.; Cai, H.; Liang, J.; Song, F.; Hou, C.; Huang, S.; et al. N6-methyladenosine demethyltransferase FTO-mediated autophagy in malignant development of oral squamous cell carcinoma. Oncogene 2021, 40, 3885–3898. [Google Scholar] [CrossRef]
- Ding, L.; Wang, R.; Zheng, Q.; Shen, D.; Wang, H.; Lu, Z.; Luo, W.; Xie, H.; Ren, L.; Jiang, M.; et al. circPDE5A regulates prostate cancer metastasis via controlling WTAP-dependent N6-methyladenisine methylation of EIF3C mRNA. J. Exp. Clin. Cancer Res. 2022, 41, 187. [Google Scholar] [CrossRef]
- Sun, H.-D.; Xu, Z.-P.; Sun, Z.-Q.; Zhu, B.; Wang, Q.; Zhou, J.; Jin, H.; Zhao, A.; Tang, W.-W.; Cao, X.-F. Down-regulation of circPVRL3 promotes the proliferation and migration of gastric cancer cells. Sci. Rep. 2018, 8, 10111. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).