Abstract
During evolution, gene duplications lead to a naturally increased gene dosage. Duplicated genes can be further retained or eliminated over time by purifying selection pressure. The retention probability is increased by functional diversification and by the acquisition of novel functions. Interestingly, functionally diverged paralogous genes can maintain a certain level of functional redundancy and at least a partial ability to replace each other. In such cases, diversification probably occurred at the level of transcriptional regulation. Nevertheless, some duplicated genes can maintain functional redundancy after duplication and the ability to functionally compensate for the loss of each other. Many of them are involved in proper embryonic development. The development of particular tissues/organs and developmental processes can be more or less sensitive to the overall gene dosage. Alterations in the gene dosage or a decrease below a threshold level may have dramatic phenotypic consequences or even lead to embryonic lethality. The number of functional alleles of particular paralogous genes and their mutual cooperation and interactions influence the gene dosage, and therefore, these factors play a crucial role in development. This review will discuss individual interactions between paralogous genes and gene dosage sensitivity during development. The eye was used as a model system, but other tissues are also included.
1. Introduction
The number of genes is organism-specific, with the budding yeast Saccharomyces cerevisiae having a total of ~6275 genes [1], the fruit fly ~13,600 genes [2], the chicken ~20–23,000 [3], the dog ~19,000 [4], the pig ~22,342 [5], the mouse ~30,000 [6], and the human ~20,000–25,000 genes. The gene dosage represents the number of copies of particular genes present in the genome. The number of gene copies can be naturally increased by duplication. The creation of an extra gene copy leads to the initial amplification of the gene dosage, which is either further maintained or eradiated. Duplication events include small-scale duplications (SSD), such as tandem or segmental gene duplications, and whole-genome duplications (WGD) that duplicate all the genetic information present in the genome [7]. Genes that have evolved through gene duplication within the same genome are classified as paralogs [8]. Examples of entire gene families being duplicated during evolution include the zinc finger transcription factor (ZNF) subfamily KRAB-ZNF on human chromosome 19 [9], ~200 G-protein-coupled receptors (GPCRs) in humans [10], the serine protease inhibitor (serpin) family [11,12], the mouse and human fibroblast growth factor family (FGF) [13], and the olfactory receptor gene (OR) superfamily [14,15]. Interestingly, during early vertebrate evolution, the ancestral vertebrate underwent two rounds (2R) of WGD [16]. Teleost fish underwent an additional, fish-specific third round (3R) of WGD [17,18]. Multiple rounds of gene duplication allowed the creation of innovative functions during evolution [19].
Duplicated genes are initially redundant in function. Functional redundancy means that duplicates have the same function, with none or minor phenotypic consequences in the case that one copy is eliminated [20]. The remaining paralogous gene is sufficient to compensate for the loss of the second paralog. Therefore, redundant duplicates provide an ideal source of genetic material that can be used as the origin of new genes [21]. Duplicated genes can follow different fates after the initial duplication (Figure 1). Duplicated genes can be further maintained when it is advantageous for the organism, or eliminated through selective pressure during evolution. Maintained duplicated genes can retain functional redundancy or become functionally divergent. Dosage amplification and back-up compensation contribute to the maintenance of functionally redundant gene copies. Dosage amplification is characterized by an increased number of gene copies, and back-up compensation is characterized by the ability of a paralogous gene to replace the loss of its paralogous partner [21]. However, the acquisition of novel functions increases the probability of retaining duplicated genes [22].
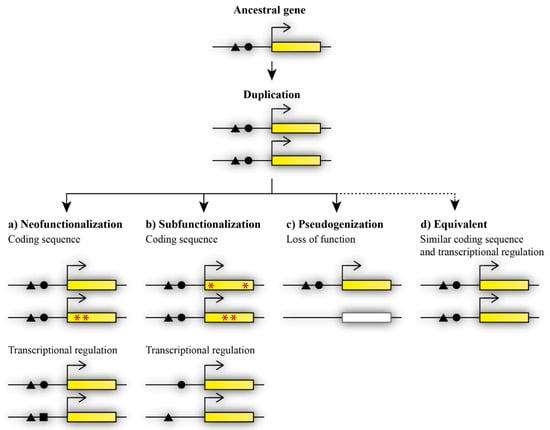
Figure 1.
Possible fates of paralogous genes after duplication. Duplicated genes can acquire divergent functions via (a) neofunctionalization, in which one duplicate retains the ancestral gene function while the other duplicate acquires a novel function, and the acquisition of the novel function occurs through mutations in the gene coding sequence (indicated by red asterisks) or changes in transcriptional regulation (black square), or (b) subfunctionalization, in which mutations change the functions of both duplicates at the level of coding sequence (red asterisks) or transcriptional regulation (missing black triangle or circle), therefore causing the presence of both copies to be essential for the maintenance of the ancestral function. Other possible fates of duplicated genes are (c) loss of function during the pseudogenization process, or, less frequently occurring, (d) maintained equivalent functions.
Functional diversification may occur via neofunctionalization or subfunctionalization and can contribute to the maintenance of both paralogous genes [21]. During neofunctionalization, a paralogous gene acquires a new function that was not present in the ancestral genome [21]. The novel function is achieved by the accumulation of mutations that may occur in the coding or regulatory sequence in one of the duplicated genes. The other duplicate thus retains the ancestral function (Figure 1a). Subfunctionalization, as previously described via the duplication–degeneration–complementation (DDC) model, results in the division of the ancestral function between paralogous genes [23]. Each duplicate retains part of the ancestral gene’s function. Mutations may occur in the coding or regulatory sequence at different sites of each duplicate and thus lead to changes in the coding sequence or the spatiotemporal diversification of expression between the duplicated genes [24]. The regulation of gene expression, expression patterns, and changes at the protein level play an important part in the acquired divergent functions after duplication [25,26,27]. In contrast, pseudogenization contributes to the eradication of the redundant copy (Figure 1c). One of the duplicates becomes a null allele, and thus loses its function after duplication [7]. In this case, the extra copy was not maintained, probably due to it not being beneficial for the organism and not improving fitness or specific features of the organism. In general, the loss of an extra copy is more frequent than the maintenance of a functionally redundant copy [20]. Nevertheless, some duplicated genes can maintain initial redundancy during evolution (Figure 1d).
Paralogous genes with a retained function observed within one organism can perform redundant and/or divergent functions. Functional redundancy and divergence between duplicates may vary in different tissues and organs within the same organism [28,29,30,31,32,33]. Moreover, some duplicated genes can perform redundant functions during development, but acquire divergent specific roles in adulthood [34].
The required threshold level may be specific for each tissue and organ, leading to a different sensitivity to the overall amount of the gene product during development. A lower amount of the gene product (below the threshold) can disrupt the proper development and fitness of the developing embryo.
Haploinsufficient genes require the presence of both copies. The loss of one copy reduces the required gene dosage and is linked with many human disorders. For example, the haploinsufficiency of T-box transcription factor 5 (TBX5) leads to congenital heart disease [35]; the haploinsufficiency of proline-rich 12 (PRP12) causes neurodevelopmental and eye diseases [36]; SHANK3 mutations predispose to autism [37]; SETD1A is linked with neurodevelopmental disorders [38]; a SOX5 haploinsufficiency is linked with neurodevelopmental disorders, intellectual disabilities, and language delays [39]; a BAZ1B haploinsufficiency is linked with neurodevelopmental disorders [40]; and the haploinsufficiency of PUM1 causes developmental delays and seizures [41].
In this review, I provide illustrative examples of a functional overlap between paralogous genes involved in embryonic development, with an emphasis on the gene dosage requirements and sensitivity leading to more or less severe alterations in proper embryonic development. The amount of the gene product may be crucial for the development of particular tissues and organs. The sensitivity of particular tissues to the gene dosage and the functional equivalency of paralogous genes were examined by combinations of single and compound losses of paralogous gene functions. The degree of retained functional equivalency was also studied using a series of replacement experiments. Individual sections of this review will discuss the degree of the retained functional equivalency of paralogous genes, the maintained redundancy between paralogous genes, the gene dosage requirements, the sensitivity of particular tissues/organs and processes during embryonic development, and finally, the acquired divergent functions of duplicated genes.
2. Retained Functional Equivalency between Paralogous Genes
The functional equivalence between paralogous genes during development was examined using a series of experiments where genes were replaced by their paralogs. The results of the studies revealed that many paralogous genes retained a certain level of functional equivalence and were able to replace each other. Functional equivalence was observed for paralogous genes that function redundantly, but also for paralogous genes with divergent functions. The acquisition of novel expression sites after the initial duplication increased the probability of gene retention and allowed for the acquisition of novel functions, while the functional interchangeability of duplicated genes was mostly maintained [22].
2.1. Interchangeable Paralogs with Divergent Functions
Many paralogous genes that had evolved distinct functions maintain a high degree of functional equivalency and are able to replace each other. As an example, the Myc family of the cellular oncogenes N-myc and c-myc are essential for mouse embryonic development, cellular growth, and differentiation. c-myc and N-myc have broad divergent expression patterns that change during development. Mice with a c-myc coding sequence replaced by the N-myc coding sequence were viable with normal fertility. Normally, the aberrant expression of c-myc leads to embryonic lethality. The N-myc expressed from the c-myc locus that properly mimicked c-myc expression was functionally complementary and regulated similarly to c-myc. The differences between the c-myc and N-myc paralogous genes are probably caused by different transcriptional regulation, leading to distinct specific expression patterns [26]. Divergence driven at the transcriptional level enables paralogs to functionally replace each other after substitution.
As another example, the mouse Engrailed genes Engrailed-1 and Engrailed-2 have different expression patterns, and their deletion leads to divergent phenotypes. The loss of Engrailed-1 leads to death after birth with the absence of the major part of the midbrain–hindbrain, while the loss of Engrailed-2 causes alterations in the cerebellum, without affecting mouse viability. Engrailed-1 with the coding sequence replaced by the Engrailed-2 coding sequence is able to compensate for and rescue defects in Engrailed-1 mutants. The ability to rescue a mutant phenotype by the replacement of paralogous genes suggests that their divergent functions are due to differences in the expression pattern [42]. Axin and Axin2, which are essential components of Wnt signaling, also exhibit functional interchangeability. The loss of Axin leads to early embryonic lethality. The deletion of the paralogous gene Axin2 leads to a different phenotype; mice lacking Axin2 are viable with craniofacial defects. However, the expression of Axin2 under the Axin promoter reveals a functional equivalency between Axin and Axin2, despite their divergent functions in normal conditions. Axin2 expressed from the Axin locus rescued early embryonic lethality in Axin-null mice. The absent redundancy between Axin and Axin2 can be a result of their different expression patterns leading to divergent functions [43]. Taken together, different functions of interchangeable paralogous genes are suggested as a consequence of the differing regulation of gene expression and acquired divergent expression patterns.
Interchangeable functions were also observed between coat protein complex II (COPII) components. Coat protein complex II (COPII) vesicles normally mediate transport from the endoplasmic reticulum to the Golgi. The core COPII components Sec23a and Sec23b acquired distinct functions after duplication. Alterations to their function thus resulted in distinct phenotypes. In humans, the loss of SEC23A leads to an abnormal skeletal phenotype due to the altered secretion of collagen, while the loss of SEC23B affects the development of red blood cells. SEC23B is highly expressed in human bone marrow, but in mice, it is broadly expressed in the pancreas. The Sec23a coding sequence placed into the mouse Sec23b locus rescued mortality and pancreatic abnormalities in Sec23b-null mice. The Sec23a coding sequence expressed from the Sec23b locus was sufficient to rescue the phenotype in Sec23b-null mice, but was not sufficient to rescue the phenotype in Sec23a-null mice, suggesting that the exact expression pattern cannot be recapitulated from a different locus [25].
Next, the coat protein complex II (COPII) components Sec24c and Sec24d play an important role during mouse development. The loss of Sec24c causes very early embryonic lethality before the eight-cell stage, while Sec24d deficiency results in early embryonic lethality around E7.5. Embryonic lethality caused by the loss of Sec24c can be rescued by the Sec24c allele with a replacement of 90% of the C-terminal domain of the paralogous Sec24d coding sequence expressed from the Sec24c locus. The recombinant allele expressed from the Sec24c locus is sufficient to compensate for the loss of Sec24c, but insufficient to rescue the phenotype in Sec24d-null mice. Probably, the exact expression pattern has not been fully recapitulated when expression occurs in a different locus, or the residual Sec24c sequence may impact the ability of functional replacement. In addition, acquired paralog-specific functions could be an explanation for this insufficient compensation. Overall, the experiments showed that Sec24c and Sec24d maintained functional equivalency and can compensate for each other when they are expressed from the locus of the replaced paralogous gene. The divergence in the Sec24c/d protein function is thus probably driven by different tissue- or stage-specific expression [44]. These experiments suggest that the expression pattern can be recapitulated only from the locus of the replaced paralogous gene and that functionally divergent paralogous genes can maintain functional redundancy after duplication even when they normally perform divergent functions.
Additionally, the paired box (Pax) protein coding genes Pax2 and Pax5 have divergent expression, with the exception of CNS. Both have overlapping expression in the midbrain–hindbrain boundary. The deletion of Pax2 or Pax5 leads to different phenotypes. The loss of Pax2 causes an absence of a posterior midbrain and cerebellum, while the deletion of Pax5 results in mild defects in the brain. Nevertheless, both proteins have similar biochemical functions, and Pax5 expressed from the Pax2 locus can substitute for Pax2 functions in the developing ear, eye, and urogenital system. Normally, Pax5 is not expressed in these tissues. The divergent functions of these paralogous genes are suggested to be a consequence of different timing and levels of their expression. Pax2 expression starts earlier than expression of Pax5 during development [45]. The timing of gene expression thus also plays an important role in the exact function of paralogous genes. Biochemical protein properties were probably maintained, and the paralogous gene was thus able to replace and compensate for the phenotypical consequences after the loss of the second paralog.
2.2. Common and Unique Functions of Paralogous Genes
Functionally redundant paralogous genes were mostly able to replace each other. The members of the Cdx family Cdx1 and Cdx2 have a functionally redundant function. Cdx2 as a substitute for Cdx1 in Cdx1-null mice was able to compensate for Cdx1 functions. Knock-in mutant mice did not exhibit skeletal defects, the expression of HOX genes proceeded normally, and the anterior–posterior vertebral patterning was not altered [46]. Cdx1 and Cdx2 were able to replace each other without phenotypic consequences after the replacement, probably due to the maintained functional redundancy.
Nevertheless, the replacement of the orthodenticle homeobox (Otx) protein-coding gene Otx1 by Otx2 and vice versa exhibited a high degree of similarity, but these genes cannot compensate for each other to the full extent. Otx2 replaced by Otx1 is able to function normally in gastrulation and in the rostral neuroectoderm, but not in rostral brain development before the normal onset of Otx1 expression during development. Replacement led to the loss of anterior structures, including eyes. This indicated that Otx2 developed novel functions in the anterior neuroectoderm that are not present in Otx1 [47]. The replacement of Otx paralogous genes was also performed between different species. Human OTX2 expressed from the Otx1 locus was able to rescue defects in Otx1-null mice. The mutant mice did not develop epilepsy, and corticogenesis proceeded normally [48]. Additionally, the human OTX1 coding sequence expressed under the transcriptional control of Otx2 had a similar expression pattern as Otx2; the mice were phenotypically normal and fertile without defects in the hindbrain and midbrain, and the brain developed normally after the replacement [49]. Paralogous genes with redundant functions were mostly able to replace each other. However, paralogous genes were sometimes not able to replace each other to the full extent, even despite a high degree of functional similarity.
Insufficient compensation was observed in the case that one paralog acquired unique functions. Pax3 and Pax7 have partially overlapping expression patterns, which, despite the partial overlap, lead to different phenotypic consequences after their deletion. To investigate their functions, Pax3 was replaced by Pax7. Pax7 was able to substitute for the functions of Pax3 in the development of the neural tube, neural crest, and somite, but was not able to functionally replace Pax3 in the formation of limb muscles during development. Pax3 has a unique role in the migration, delamination, and proliferation of muscle precursor cells that cannot be rescued by its paralog Pax7 [50]. The transforming growth factor β (Tgfb) isoforms Tgfb1 and Tgfb3 are important for proper embryonic development. The loss of Tgfb3 results in secondary palate alteration. To test their functional equivalency, Tgfb1 was inserted instead of the coding sequence of exon 1 in the Tgfb3 gene. The replacement of Tgf-β3 by Tgf-β1 only partially saved the epithelial fusion defect in Tgfb3-null mice. The Tgf-β3 isoform has a specific role in palatal epithelium formation that cannot be compensated for by Tgf-β1 [51].
A similar example comes from Delta-like canonical Notch ligands (Dll); Dll1 and Dll4 have partially redundant functions in myogenesis, fully redundant functions in the maintenance of retinal progenitors, and divergent functions in somitogenesis. Dll4 expressed from the Dll1 locus in Dll1-null mice is able to replace Dll1 in the retina, but is not able to mediate the proper segmentation of the embryo. Only Dll1 is sufficient for proper embryo segmentation; therefore, Dll4 cannot replace the Dll1 functions during embryonic segmentation. The different functions were probably caused by different cis-inhibitory properties [31]. Knock-in experiments revealed a partial interchangeability between the members of the Pax family genes Pax6, Pax6(5a), and Pax2. Pax6 can be replaced by Pax6(5a) or Pax2 during brain development. The genes have a similar binding specificity and a similar paired domain. However, Pax6 has acquired a unique role in eye development and cannot be replaced by Pax6(5a) or Pax2. The substitution of Pax6 by Pax6(5a) or Pax2 leads to the failure of lens and retinal development [52]. The paralogous genes involved in eye development and their mutant phenotypes are summarized in Table 1.

Table 1.
Overview of paralogous genes involved in eye development.
Paralogous genes were not sufficient to compensate for isoform-specific functions performed by the second paralog. Paralogs retained only partial functional equivalency. The loss of a paralogous gene and its replacement by a second paralog thus led to mild or severe phenotypic consequences.
3. Redundant Functions of Paralogous Genes
Paralogous genes are involved in many essential developmental processes and play a crucial role in the proper development of embryonic tissues and organs. Functionally redundant paralogous genes have maintained a certain level of functional redundancy, and can thus at least partially compensate for the loss of each other. The possible types of cooperation between paralogous genes are shown in Figure 2. Fully redundant paralogs are characterized by their ability to compensate for the loss of the second paralog to the full extent. As examples, studies will be mentioned in the following section.
Dll1 and Dll4 act redundantly in the maintenance of retinal progenitor cells and can functionally replace each other. A fully redundant function was also observed in the maintenance of crypt progenitor cells in the adult small intestine [31]. Hoxa9 and Hoxd9 perform fully redundant functions in the formation of the humeral head. Hoxa9/Hoxd9 compound mutant mice displayed alterations in the shape of the humeral head that were not observed in single Hoxa9 and Hoxd9 mutants. Hoxa9 fully compensated for the loss of Hoxd9 in the humeral head and vice versa [68]. The Lef/Tcf transcription factors Lef1, Tcf7, Tcf7l1, and Tcf7l2 are important during lung development. A complete redundancy in epithelial progenitor cells during lung development was observed in cases when two or more out of the four paralogous genes were present [69]. The histone deacetylases Hdac1 and Hdac2 have fully redundant functions in maintaining proper chromatin structures that are important during developmental processes [70]. The Nk6 homeobox (Nkx6) genes Nkx6.1 and Nkx6.2 have fully redundant roles in α-cells during pancreatic development [29,30].
The Sall-like (Sall) protein-coding genes Sall1 and Sall4 play redundant roles during neural tube development. Sall1/Sall2 double knockout mice have no obvious alterations in their neural tube development, and Sall4 was able to compensate for the loss of its paralogous genes in the neural tube [71]. The nuclear Dbf2-related (Ndr) family of serine/threonine protein kinases genes Ndr1 and Ndr2 function redundantly in cardiac looping and during somite genesis, and can compensate for the loss of each other. Even a single allele of Ndr1 or Ndr2 is sufficient for proper heart development and somitogenesis [72]. This is consistent with the fully redundant function between paralogous genes and their ability to compensate for the loss of each other.
Membrane-associated phosphatidylinositol transfer proteins (Pitpnm) have also maintained a redundant function. Pitpnm1 is expressed in the inner ear, and its deletion does not cause hearing defects. Probably, the loss of Pitpnm1 is compensated for by the paralogous genes Pitpnm2 and Pitpnm3 [73]. The transforming growth factor β (Tgf-β) proteins Tgf-β1 and Tgf-β2 act redundantly in secondary palate development in a strain-dependent manner. Tgfb1 and Tgfb2 single knockouts have no palate defects in the C57BL/6J background, suggesting that both paralogous genes function redundantly in the C57BL/6J strain. Defects in secondary palate formation were observed in the mixed 129 and Black Swiss strain, suggesting a strain-dependent phenotype in secondary palate development [74]. The maternally imprinted minor splicing factors Zrsr1 (also known as U2af1-rs1) and Zrsr2 (also known as U2af1-rs2) are encoded by the Zrsr1 and Zrsr2 paralogous genes. Both genes are essential for zygotic genome activation and both act in a redundant manner. At least one maternal Zrsr2 or one paternal Zrsr1 allele is necessary and sufficient for early development [75].
In addition, the homeobox genes Meis1 and Meis2 exhibited redundant roles in the lens placode during lens formation [60] (Figure 2a) and in the pool of retinal progenitor cells [61]. The acetyltransferases CBP and p300 displayed redundant roles in lens induction, and even a single functional allele of CBP or p300 was sufficient for lens formation [53]. The fibroblast growth factor receptor genes Fgfr1, Fgfr2, Fgfr3, and Fgfr4 play a redundant role in lens fiber differentiation [55]. Six3 and Six6 redundantly regulate the maintenance of multipotent neuroretinal progenitors [67].
Fully redundant paralogous genes can compensate for the loss of each other without phenotypic consequences during embryonic development. The deletion of one paralog thus does not lead to phenotypic alterations due to the presence of and functional compensation by the other paralog (Figure 2a).


Figure 2.
Functional cooperation between paralogous genes. (a) Paralogous genes can maintain functional redundancy and the ability to compensate for the loss of each other without phenotypic consequences. Meis1 can compensate for the lack of Meis2 and vice versa. Only the combined deletion of paralogous genes results in severe phenotypic alterations. The combined loss of Meis1 and Meis2 leads to arrested lens development [60]. (b) Paralogs can act additively according to the quantitative model of function. The decreased number of functional alleles causes gradual worsening of the mutant phenotype. Lack of two Onecut1/Onecut2 alleles in the mouse retina causes a dramatically decreased number of horizontal cells in any combination. One remaining Onecut1 or Onecut2 allele, similarly to the combined loss of Onecut1 together with Onecut2, results in complete loss of horizontal cells [62]. (c) A synergistic interaction results in a mutant phenotype that exceeds expectations from observations of single knockouts. Hfs1-null mice have normal testes, in contrast to Hsf2-null mice with decreased testis size. Surprisingly, additional deletion of Hsf1 in Hsf2-null mice causes an even more decreased testis size [76].
Figure 2.
Functional cooperation between paralogous genes. (a) Paralogous genes can maintain functional redundancy and the ability to compensate for the loss of each other without phenotypic consequences. Meis1 can compensate for the lack of Meis2 and vice versa. Only the combined deletion of paralogous genes results in severe phenotypic alterations. The combined loss of Meis1 and Meis2 leads to arrested lens development [60]. (b) Paralogs can act additively according to the quantitative model of function. The decreased number of functional alleles causes gradual worsening of the mutant phenotype. Lack of two Onecut1/Onecut2 alleles in the mouse retina causes a dramatically decreased number of horizontal cells in any combination. One remaining Onecut1 or Onecut2 allele, similarly to the combined loss of Onecut1 together with Onecut2, results in complete loss of horizontal cells [62]. (c) A synergistic interaction results in a mutant phenotype that exceeds expectations from observations of single knockouts. Hfs1-null mice have normal testes, in contrast to Hsf2-null mice with decreased testis size. Surprisingly, additional deletion of Hsf1 in Hsf2-null mice causes an even more decreased testis size [76].


Nevertheless, paralogous genes can acquire specific functions during evolution and maintain only a partial functional redundancy with their paralog. During embryonic development, partial functional redundancy was observed among many paralogous genes. Some examples include the following: Dll1 and Dll4 have a partially redundant function during embryonic myogenesis [31]. The dishevelled proteins (Dvl) Dvl1, Dvl2, and Dvl3 function partially redundantly during heart development [28]. The Sost domain-containing (Sost) paralogous genes Sost and Sostdc1 play partially redundant and complementary roles during limb development, despite their different non-overlapping expression patterns [77]. Odd-skipped related transcription factor 1 (Osr1) acts redundantly with Osr2 in the formation of synovial joints. The deletion of Osr2 results in only subtle defects in synovial joint development [78]. The immunoglobulin-like cell adhesion proteins nectin1 and nectin3 showed partially redundant functions during tooth development [79].
As another example, the Onecut transcription factors Onecut1 and Onecut2 have a partially redundant function in pancreas organogenesis and in the subsequent steps of pancreatic endocrine differentiation [32] Another function of Onecut transcription factors is discussed in Section 4.1 and Section 5. The transmembrane proteins TMEM120A and TMEM120B function at least partially redundantly during adipocyte differentiation [80]. Hoxa5 and Hoxb5 showed partial redundancy during lung morphogenesis [81]. Hox6 genes have a high degree of functional redundancy in pancreatic organogenesis [82], and the RNA polymerase III subunits Polr3g and Polr3gl are functionally redundant during embryonic development and can partially compensate for each other [83]. Partially redundant paralogous genes perform both common and divergent functions. Therefore, partially redundant paralogs can only partly rescue a phenotype after the loss of the second paralogous gene. Specific functions cannot be compensated for by the other paralogous gene. In general, acquired novel functions are suggested to increase the probability of gene retention, and thus the probability of escape from purifying selection pressure.
4. Gene Dosage Sensitivity and Allelic Interactions
Embryonic development can be dramatically influenced by paralogous genes that take part in many developmental processes, and their loss may have severe consequences. The alteration of individual alleles or even the combined loss of paralogous genes changes the level of gene products, which can be critical for the development and maintenance of particular tissues and organs. The required threshold level may differ for individual tissues and organs, leading to different gene dosage sensitivities. The gene dosage required for normal function is thus dependent on the number of functional alleles of paralogous genes. Individual alleles of paralogous genes exhibit different levels of functional redundancy and cooperation, depending on a qualitative or quantitative model. In the quantitative model, they may act additively or even exhibit a synergistic interaction, where the phenotype of the compound mutant is worse than the phenotype that can be expected from individual single knockouts.
4.1. Additive Functions
A functional redundancy was observed among Dlx gene pairs, Dlx1–Dlx2 and Dlx5–Dlx6. The Dlx5 and Dlx6 paralogous genes exhibited a quantitative model of function in craniofacial morphogenesis. The alleles showed a high degree of functional equivalency and functioned additively. The deletion of Dlx5 or Dlx6 had the same phenotypic consequences; both resulted in an alteration to the mandibular arch. Double Dlx5/Dlx6 heterozygotes also exhibited alterations to the mandibular region. The loss of two Dlx5/Dlx6 alleles in any combination had similar phenotypic consequences. The deletion of three Dlx5/Dlx6 alleles led to a markedly reduced or lost proximal dentary structure, whereas the loss of all four alleles caused a transformation of the lower jaw structures to a maxillary identity. The normal pharyngeal patterning without alterations requires a threshold activity of Dlx5 and Dlx6; at least three functional alleles of Dlx5/Dlx6 must be present. Dlx5 and Dlx6 contribute proportionally and additively to craniofacial morphogenesis [84].
Gene dosage sensitivity was also observed in the developing mouse retina. The functionally redundant Onecut transcription factors Onecut1 and Onecut2 function additively in the context of the horizontal cell population (Figure 2b). A single deletion of Onecut1 or Onecut2 results in a markedly reduced number of horizontal cells in the mouse retina. A compound Onecut1/Onecut2 deletion leads to the complete loss of horizontal cells. At least three functional alleles are required for the normal development and maintenance of horizontal cells. A loss of two out of the four functional alleles in any combination leads to a dramatic decrease in horizontal cells. A lack of three or all four alleles results in the complete loss of horizontal cells. Therefore, only one functional Onecut1 or Onecut2 allele is not able to rescue the phenotype, and the horizontal cells will be depleted [62].
Next, a functional redundancy in a dosage-sensitive manner was revealed between Hoxa10, Hoxc10, and Hoxd10 during kidney development. The combined deletion of any three alleles did not alter normal kidney development. Defects in the kidney development were observed after the deletion of four alleles. The deletion of five alleles led to stronger defects. A lack of six alleles demonstrated the most severe alterations during kidney development; triple knockout mice died after birth. The indistinguishable mutant phenotypes indicate an additive function between the Hox10 alleles. Kidney development is thus sensitive to the overall Hox10 gene dosage, requiring the presence of at least three functional alleles for normal kidney development [85]. In general, additively functioning alleles of paralogous genes show a high degree of functional equivalency and exhibit a quantitative model of function (Figure 2b). Particular tissues are sensitive to gene dosage, and thus require a certain number of functional alleles. A gradual decline in functional alleles leads to a gradually worsened phenotype. A drop below the threshold level, which can be different for particular tissues and paralogous genes, has dramatic phenotypic consequences.
Moreover, additive functions between paralogous genes were also observed in Lef/Tcf transcription factors during lung development. Lef/Tcf factors function in the specification of epithelial progenitors. Interestingly, in the presence of at least two out of the four Lef/Tcf transcription factors, Lef/Tcf factors functioned redundantly. Two remaining paralogous transcription factors were sufficient to fully compensate for the loss of their paralogs. Triple Lef/Tcf knockouts displayed varying phenotypes, and quadruple knockouts resulted in underdeveloped lungs and minimal activity of epithelial progenitors. Lef/Tcf factors acted redundantly, but after the initial loss of two, the paralogous Lef/Tcf factors acted additively in the specification and maintenance of epithelial progenitor cells. The redundant and additive functions were dependent on the overall gene dosage. It was also shown that the individual Lef/Tcf transcription factors were not equal. A single remaining Lef1 or Tcfl2 provided partial function in epithelial progenitors, while a single functional Tcf7 or Tcf7l1 provided full function [69]. This research led to the interesting discovery that additive functions can also be observed between non-equivalent paralogous genes.
4.2. Synergistic Interaction between Paralogous Genes
Heat-shock factors (Hsf) displayed synergic functions. Hsf1-null mice had normal spermatogenesis, while Hsf2-null mice had smaller testes and a mild impairment of fertility. Defects in compound mutants were unexpectedly more severe than in single Hsf2-null mice (Figure 2c). The deletion of both Hsf1/Hsf2 factors caused defects in spermatogenesis that led to male sterility. Hsf activity is thus essential for normal spermatogenesis and fertility [76]. Additionally, an examination of Hsf4-null mice showed that the testes were not affected by an Hsf4 deletion [58]. The loss of Hsf4 in the testis was probably compensated for by other paralogous genes. Interestingly, the paralogous Hsf genes acquired a stage-specific expression in the lens. Hsf1 and Hsf2 are expressed in the fetal lens. The deletion of Hsf1 or Hsf2 had no phenotypic consequences in the lens, probably due to compensation by a paralogous gene. In contrast to Hsf1 and Hsf2, Hsf4 is predominantly expressed postnatally and regulates the expression of αβ-crystallins [59]. The deletion of Hsf4 led to cataracts and abnormal lens fibers [58]. The Hsf family member Hsf3 acquired unique functions in mice to protect the cell from stress by activating nonclassical heat-shock genes. In humans, Hfs3 has become a pseudogene [86].
Synergistic interactions were also observed during axial skeleton development. A lack of Pax1 or Pax9 resulted in divergent phenotypes. The deletion of Pax1 resulted in morphological alterations to the axial skeleton, while the deletion of Pax9 did not cause axial skeleton defects. Nevertheless, the additional deletion of Pax9 in Pax1-null mice resulted in a more severe phenotype. Compound Pax1/Pax9 mice lost their medial derivates of sclerotomes, vertebral bodies, invertebral discs, and proximal parts of the ribs. Cooperation between Pax1 and Pax9 is dosage-dependent; the deletion of three functional alleles causes intermediate phenotypes. The worsened phenotype in Pax1-null mice after the additional deletion of Pax9 revealed a synergistic interaction between Pax1 and Pax9 [87]. A synergistic interaction was also observed in the Hox3 gene cluster. Single Hoxa3 and Hoxd3 mutants had no defects in the formation of their cervical vertebrae. The deletion of Hoxb3 caused slight defects in the formation of cervical vertebrae with low penetrance. Surprisingly, the combined loss of Hoxa3/Hoxd3 or Hoxb3/Hoxd3 caused a loss of the entire atlas, suggesting a synergic function of Hox3 genes. Hox3 alleles exhibit dosage-dependent interactions and interact in a quantitative manner. The removal of any of the three Hox3 alleles led to the partial loss of the atlas, whereas the loss of four alleles caused a more severe phenotype, the loss of the entire atlas. Compound Hoxa3/Hoxd3 and Hoxb3/Hoxd3 mutants exhibited the same cervical vertebrae alteration as Hoxa3+/−/Hoxb3+/−/Hoxd3−/− triple mutants. Indistinguishable defects observed after the loss of different allele combinations indicated an equivalent function of the Hox3 paralogous genes in the cervical vertebrae [88]. A synergistic interaction is characterized by an unexpected mutant phenotype that cannot be expected from observations of single knockouts (Figure 2c). The additional deletion of a paralogous gene in a single knockout leads to more severe phenotypic consequences, despite the fact that the single knockout of the additionally deleted paralogous gene does not have phenotypic consequences.
As another example of synergistic interaction, I can mention Dvl genes. The deletion of individual Dvl1, Dvl2, and Dvl3 genes results in divergent alterations. A lack of Dvl1 leads to abnormalities in social interactions and altered sensorimotor gating. A lack of Dvl2 causes cardiac outflow tract abnormalities and rib and vertebral malformations with incomplete penetrance. The loss of Dvl3 leads to defects in the inner ear, neural tube defects, and cardiac outflow tract abnormalities causing perinatal lethality. Nevertheless, Dvl genes have overlapping expression patterns and exhibit functional redundancy even despite their distinct mutant phenotypes. The additional loss of Dvl paralogous genes causes more severe phenotypes, suggesting gene-dosage sensitive redundant roles of Dvl genes. A single Dvl knockout did not display neural closure defects. The combined loss of three or all four alleles of Dvl1/Dvl2 or Dlv2/Dvl3 resulted in neurulation defects. Neurulation in the compound Dvl1/Dvl3 mutant proceeded normally. Probably, the alleles are not functionally equivalent. Gene dosage sensitivity was also observed in the organ of Corti. Single Dvl2 and Dvl3 heterozygotes had normal development of the organ of Corti, whereas double heterozygotes displayed alterations to the development of the organ of Corti. The additional deletion of the Dvl2 allele in Dvl3-null mice worsened the phenotype. A dosage-sensitive function was also observed in cardiac development. Double Dvl2/Dvl3 heterozygotes exhibited cardiac defects, in contrast to single Dvl2 and Dvl3 heterozygotes with normal cardiac development. A more severe phenotype was observed after the deletion of three Dvl2/Dvl3 alleles. A non-equivalent function was observed in rescue experiments. An extra copy of Dvl1 was not able to restore cardiac defects in Dvl2-null mice, whereas an additional copy of Dvl1 and Dvl2 was able to compensate for the loss of Dvl3 during heart development [28]. Synergistic interactions can thus be observed between equivalent, but also between non-equivalent, paralogous genes.
5. Functionally Divergent Paralogs
Naturally, there are a number of instances in which the paralogous genes have acquired distinct functions. For example, the loss of the homeobox (HOX) gene Hoxc-4 does not affect the cervical vertebrae, in contrast to its paralogous genes Hoxa-4, Hoxb-4, and Hoxd-4, but leads to abnormalities in the thoracic vertebrae [89]. Hoxa10 and Hoxd10 act independently in the regulation of lumbar and sacral axial patterning. Mutations in Hoxa10 are the cause of lumbar vertebral transformations, while Hoxd10 mutations affect the sacral vertebrae [90]. H3K4 methyltransferase MLL3 (also known as KMT2C, lysine methyltransferase 2C) diverged from its paralog MLL4 (also known as KMT2D, lysine methyltransferase 2D). MLL3 is essential for lung maturation, while MLL4 is required for the initiation of gastrulation via the regulation of anterior visceral endoderm migration [91].
As another example, the Onecut transcription factor Onecut1 functionally diverged from its paralogous genes Onecut2 and Onecut3. Only Onecut1 is required for the specification and morphogenesis of the pancreas and endocrine differentiation. Moreover, Onecut2 and Onecut3 are not required for gut development and enteroendocrine differentiation, in contrast to Onecut1 [32]. Serine protease (Prss) 55, in contrast to its paralog Prss51, is essential for male fertility, sperm migration, and binding to the zona pellucida [92]. Sost and Sostdc1 diverge in the majority of organ systems, including the nervous, cardiovascular, musculoskeletal, respiratory, reproductive, and digestive systems [77]. Hdac1 and Hdac2 have functionally diverged during evolution. Hdac2 gained a new function in the regulation of neural precursor cells in brain development during evolution, which is not present in its paralog Hdac1. A single remaining functional allele of Hdac1 is not sufficient for proper brain development, in contrast to Hdac2 [70]. The family with sequence similarity 170 member A (Fam170a) is important for male fertility, while the paralogous gene Fam170b is not essential for male fertility [93]. Paralogous genes that did not maintain functional redundancy underwent a subfunctionalization or neofunctionalization process after the initial duplication. Functional diversification is advantageous due to the increased probability of their maintenance.
Distinct functions emerged in the H3K4 methyltransferase paralogs of the SET-containing domain (Setd), Setd1a and Setd1b. Setd1a is important for gastrulation, whereas Setd1b is dispensable. Setd1b is required later in embryonic development during organogenesis [94]. The winged-helix/forkhead transcription factors (Fox) Foxa1 and Foxa2 play a redundant role during development, but have evolved distinct roles in the adult liver [34]. The TATA-binding-associated proteins (TAFs) TAF4b and TAF4 evolved different roles in regulating the maintenance and proliferation of mouse embryonic stem cells. TAF4b is highly expressed in embryonic stem cells (ESCs), supporting proliferation and progression through the cell cycle. A high proliferation rate is a specific sign of embryonic stem cells. TAF4 has the opposite effect to TAF4b and reduces the growth of ESCs. The expression of TAF4 is essential in later developmental stages [95]. The anti-silencing function 1 (Asf1) histone chaperone genes Asf1a and Asf1b acquired divergent functions during evolution. The loss of Asf1a leads to embryonic lethality, while Asf1b-null mice are viable, but exhibit decreased reproductive capacity compared to the wild type [96]. Further studies have shown that Asf1a is involved in histone H3.3 deposition in the paternal pronucleus after fertilization and regulates levels of Oct4 expression and H3K56ac. Asf1b is required for the regulation of cell proliferation in early embryos [97]. Divergent paralogous genes can be involved in distinct temporal stages during development, and thus exhibit temporal functional diversity.
Divergences in expression patterns and the performance of distinct functions have been observed between some paralogous genes. For example, HOXA cluster genes have expression patterns differing from HOXC and HOXD in the human endometrium. HOXA10 and HOXA11 regulate the differentiation of the endometrium, whereas HOXC10, HOXC11, HOXD10, and HOXD11 play a role in the regulation of endometrial proliferation [98]. The H6 homeobox genes (Hmx) Hmx1 (also known as Nkx5-3), Hmx2 (Nkx5-2), and Hmx3 (Nkx5-1) are expressed in the developing nervous system. Hmx1 acquired a developmental role differing from its paralogs Hmx2 and Hmx3 and is expressed in the eye lens and retina during development. Hmx2 and Hmx3 share a similar expression pattern in the developing central nervous system and inner eye [56]. Divergent functions of duplicated genes ensure a higher chance of their maintenance during evolution and escape from the purifying selection pressure leading to elimination of the extra gene copy over time.
Another interesting group of genes to mention are the opsins, which represent G-protein-coupled receptors (GPCRs). There are three opsins present in human cones: opsin1 long-wave-sensitive (OPN1LW, L-cones) and opsin1 medium-wave-sensitive (OPN1MW, M-cones), both localized on chromosome Xq28, and opsin 1 short-wave-sensitive (OPN1SW, S-cones), localized on chromosome 7. Cones are responsible for color vision and the recognition of red (L-cones), green (M-cones), and blue (S-cones) color. Alterations in both OPN1LW and OPN1MW lead to a recessive X-linked disorder called blue cone monochromacy (BMC). The symptoms include dramatically reduced central vision, impaired color vision, photophobia, and congenital nystagmus [63,64]. X-linked disorders are more pronounced in males due to the presence of a single X-chromosome inherited from the mother. The misalignment of opsins during the recombination of two X-chromosomes in females can lead to deuteranopia (green blindness), where the gene coding for the M pigment is replaced by the L gene, or protanopia (red blindness), where the L gene is replaced by the M gene [65]. Individuals with unaltered color vision have a single red pigment gene and one or more copies of the green pigment gene. However, only the most proximal green pigment gene is expressed [99,100]. Compared to humans, the mouse retina consists of only two types of cones, M-cones and S-cones [101].
Interestingly, paralogous genes may have redundant, but also divergent, functions within the same organism. For example, during pancreatic development, Nkx6.1 and Nkx6.2 have redundant roles in α-cell development, but distinct roles in β-cell development due to a divergent expression pattern [29,30]. Dvl1 and Dvl2 have redundant roles with Dvl3 in cardiac development, whereas their roles diverge in skeletal development. Dvl1 and Dvl2 act redundantly, and their deletion results in skeletal defects, while the loss of Dvl3 does not cause skeletal defects [28]. Dll1 and Dll4 have partially redundant functions in myogenesis and fully redundant functions in the maintenance of retinal progenitors, but have acquired divergent roles in somite segmentation [31]. The Onecut1 and Onecut2 transcription factors have redundant roles in the mouse retina, but divergent functions in endocrine differentiation and gut development [32,33]. Sometimes, redundancy/divergence between paralogous genes can switch during development. This phenomenon was observed in the case of Foxa1 and Foxa2, which play a redundant role during liver development, but acquire divergent functions in the adult liver [34].
6. Conclusions
Embryonic development can be dramatically influenced by paralogous genes and their functional cooperation. Specific developmental processes require the presence of a certain paralogous gene due to its acquired divergent role that cannot be functionally compensated for by a functionally divergent paralogous gene. Interestingly, replacement experiments have revealed the maintained ability of some functionally distinct paralogous genes to replace each other when expressed from the locus of the replaced paralog. This indicates that changes between interchangeable paralogous genes occurred at the level of transcriptional regulation. The biochemical protein properties were probably maintained, and thus sufficient replacement is enabled. Functional interchangeability was observed, for example, between the N-myc and c-myc [26], Engrailed1 and Engrailed2 [42], Axin and Axin2 [43], Sec23a and Sec23b [25], Sec24c and Sec24d [44], Pax2 and Pax5 [45], and Cdx1 and Cdx2 [46] paralogous genes. Nevertheless, some acquired unique paralog-specific functions remain irreplaceable. Irreplaceable specific functions were observed, for example, in Otx2, which acquired a specific irreplaceable role in rostral brain development [47]; Pax3, which has a unique function in muscle precursor cells [50]; Tgf-β3, which acquired a specific role in the formation of the palatal epithelium [51]; Dll1, which is required for proper embryo segmentation [31]; and Pax6, which acquired a unique role in eye development [52]. The functional diversification of paralogous genes increases the chance of their maintenance. Otherwise, the extra copy is eliminated by purifying selection pressure. A loss of function occurs during the pseudogenization process and is more common than conserved redundancy.
Despite this fact, some paralogous genes can maintain functional redundancy after the initial duplication. Functional cooperation and interactions between maintained paralogous genes influence the gene dosage that is essential for the proper development of particular tissues/organs and in developmental processes. Paralogs with a maintained functional redundancy can compensate for the absence of each other. Phenotypic consequences can be observed in cases where both paralogous genes are missing. Fully redundant functions were observed between the Dll1 and Dll4 [31]; Hoxa9 and Hoxd9 [68]; Lef/Tcf transcription factors [69]; Hdac1 and Hdac2 [70]; Nkx6.1 and Nkx6.2 [29,30]; Sall1 and Sall4 [71]; Ndr1 and Ndr2 [72]; Pitpnm1, Pitpnm2, and Pitpnm3 [73]; Tgfb1 and Tgfb2 [74]; Zrsr1 and Zrsr2 [75]; Meis1 and Meis2 [60,61]; CBP and p300 [53]; Fgfr1, Fgfr2, Fgfr3, and Fgfr4 [55]; and Six3 and Six6 paralogous genes.
Additive functions lead to a gradually worsened phenotype with a gradually decreased number of functional alleles of paralogous genes. A gradually decreased number of functional Dlx5/6 alleles leads to a gradually worsened craniofacial morphogenesis [84]; gradually reduced Onecut1/Onecut2 alleles cause a dramatically reduced number of horizontal cells in the mouse retina [62]; a gradually decreased number of functional Hoxa10, Hoxc10, and Hoxd10 alleles results in gradually worsened kidney development [85]; and a decreased number of Lef/Tcf alleles gradually alters the specification and maintenance of epithelial progenitor cells [69]. The threshold level of functional alleles is specific for particular tissues and organs, and may differ for individual paralogous genes. A decline below the threshold level results in dramatic phenotypic consequences. The loss of more than one functional Dlx5/Dlx6 allele leads to alterations in pharyngeal patterning [84], a single Onecut1/Onecut2 allele in any combination is not sufficient to prevent the complete loss of horizontal cells [62], less than three functional Hoxa10/Hoxc10/Hoxd10 alleles leads to defects during kidney development [85], and the presence of less than two out of the four Lef/Tcf transcription factors results in defects during lung development [69]. Additive functions were mostly observed for functionally equivalent paralogous genes, but non-equivalent paralogous genes may also exhibit additive functions. For example, Lef/Tcf factors function additively in the maintenance and specification of epithelial progenitor cells, but are not equal. A single remaining Lef1 or Tcfl2 factor provides partial function in epithelial progenitors, while a single functional Tcf7 or Tcf7l1 factor provides full function [69].
Next, synergistic interactions lead to unexpected phenotypes. A single knockout itself may have no phenotypic consequences, but the additional deletion in a single knockout of a paralogous gene can unexpectedly cause more severe alterations. The severity of phenotypic consequences thus cannot be predicted from the phenotypes of single knockouts. Unexpected synergistic interactions were observed between Hsf1 and Hsf2 [76], Pax9 and Pax1 [87], Hoxa3/Hoxd3 and Hoxb3/Hoxd3 [88], and Dvl paralogous genes [28]. The evolved cooperation and interactions between paralogous genes and their alleles thus play an essential role during proper embryonic development.
Although, in general terms, some predictions of paralog function can be inferred from the known functions of the original gene and the expression profile of the paralog in question, it is still difficult to predict the function of diversified paralogs with certainty. Even with the gradual accumulation of our knowledge, it is unreliable to use, for example, ortholog conjecture in the prediction of gene function [102], not to mention the dependency of paralogs or their compensatory capability for each other [103,104]. Thus, the most reliable approach still remains tedious experimental work on model organisms.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Conflicts of Interest
The author declares no conflict of interest.
References
- Liu, W.; Li, L.; Ye, H.; Chen, H.; Shen, W.; Zhong, Y.; Tian, T.; He, H. From Saccharomyces cerevisiae to human: The important gene co-expression modules. Biomed Rep. 2017, 7, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The Genome Sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, E.A.; Wayne, R.K. The canine genome. Genome Res. 2005, 15, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Sjöstedt, E.; Oksvold, P.; Sivertsson, Å.; Huang, J.; Álvez, M.B.; Arif, M.; Li, X.; Lin, L.; Yu, J.; et al. Genome-wide annotation of protein-coding genes in pig. BMC Biol. 2022, 20, 25. [Google Scholar] [CrossRef]
- Weitzman, J.B. The mouse genome. Genome Biol. 2002, 3, spotlight-20021205–20021202. [Google Scholar] [CrossRef]
- Kuzmin, E.; Taylor, J.S.; Boone, C. Retention of duplicated genes in evolution. Trends Genet. 2022, 38, 59–72. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef]
- Grimwood, J.; Gordon, L.A.; Olsen, A.; Terry, A.; Schmutz, J.; Lamerdin, J.; Hellsten, U.; Goodstein, D.; Couronne, O.; Tran-Gyamfi, M.; et al. The DNA sequence and biology of human chromosome 19. Nature 2004, 428, 529–535. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Heit, C.; Jackson, B.C.; McAndrews, M.; Wright, M.W.; Thompson, D.C.; Silverman, G.A.; Nebert, D.W.; Vasiliou, V. Update of the human and mouse SERPIN gene superfamily. Hum. Genom. 2013, 7, 22. [Google Scholar] [CrossRef]
- Hill, R.E.; Hastie, N.D. Accelerated evolution in the reactive centre regions of serine protease inhibitors. Nature 1987, 326, 96–99. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Young, J.M.; Friedman, C.; Williams, E.M.; Ross, J.A.; Tonnes-Priddy, L.; Trask, B.J. Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum. Mol. Genet. 2002, 11, 535–546. [Google Scholar] [CrossRef]
- Zhang, X.; Firestein, S. The olfactory receptor gene superfamily of the mouse. Nat. Neurosci. 2002, 5, 124–133. [Google Scholar] [CrossRef]
- Dehal, P.; Boore, J.L. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005, 3, e314. [Google Scholar] [CrossRef]
- Amores, A.; Force, A.; Yan, Y.L.; Joly, L.; Amemiya, C.; Fritz, A.; Ho, R.K.; Langeland, J.; Prince, V.; Wang, Y.L.; et al. Zebrafish HOX clusters and vertebrate genome evolution. Science 1998, 282, 1711–1714. [Google Scholar] [CrossRef]
- Christoffels, A.; Koh, E.G.; Chia, J.M.; Brenner, S.; Aparicio, S.; Venkatesh, B. Fugu genome analysis provides evidence for a whole-genome duplication early during the evolution of ray-finned fishes. Mol. Biol. Evol. 2004, 21, 1146–1151. [Google Scholar] [CrossRef]
- Holland, P.W.; Garcia-Fernàndez, J.; Williams, N.A.; Sidow, A. Gene duplications and the origins of vertebrate development. Dev. Suppl. 1994, 1994, 125–133. [Google Scholar] [CrossRef]
- Nowak, M.A.; Boerlijst, M.C.; Cooke, J.; Smith, J.M. Evolution of genetic redundancy. Nature 1997, 388, 167–171. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Cooke, J.; Nowak, M.A.; Boerlijst, M.; Maynard-Smith, J. Evolutionary origins and maintenance of redundant gene expression during metazoan development. Trends Genet. 1997, 13, 360–364. [Google Scholar] [CrossRef]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Cui, L.; Wall, P.K.; Zhang, Q.; Zhang, X.; Leebens-Mack, J.; Ma, H.; Altman, N.; dePamphilis, C.W. Expression pattern shifts following duplication indicative of subfunctionalization and neofunctionalization in regulatory genes of Arabidopsis. Mol. Biol. Evol. 2006, 23, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Khoriaty, R.; Hesketh, G.G.; Bernard, A.; Weyand, A.C.; Mellacheruvu, D.; Zhu, G.; Hoenerhoff, M.J.; McGee, B.; Everett, L.; Adams, E.J.; et al. Functions of the COPII gene paralogs SEC23A and SEC23B are interchangeable in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E7748–E7757. [Google Scholar] [CrossRef] [PubMed]
- Malynn, B.A.; de Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Shew, C.J.; Carmona-Mora, P.; Soto, D.C.; Mastoras, M.; Roberts, E.; Rosas, J.; Jagannathan, D.; Kaya, G.; O’Geen, H.; Dennis, M.Y. Diverse Molecular Mechanisms Contribute to Differential Expression of Human Duplicated Genes. Mol. Biol. Evol. 2021, 38, 3060–3077. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, S.L.; Ray, S.; Li, S.; Hamblet, N.S.; Lijam, N.; Tsang, M.; Greer, J.; Kardos, N.; Wang, J.; Sussman, D.J.; et al. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008, 4, e1000259. [Google Scholar] [CrossRef]
- Henseleit, K.D.; Nelson, S.B.; Kuhlbrodt, K.; Hennings, J.C.; Ericson, J.; Sander, M. NKX6 transcription factor activity is required for α- and β-cell development in the pancreas. Development 2005, 132, 3139–3149. [Google Scholar] [CrossRef]
- Nelson, S.B.; Schaffer, A.E.; Sander, M. The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting β-cell fate specification in Pdx1+ pancreatic progenitor cells. Development 2007, 134, 2491–2500. [Google Scholar] [CrossRef]
- Preuße, K.; Tveriakhina, L.; Schuster-Gossler, K.; Gaspar, C.; Rosa, A.I.; Henrique, D.; Gossler, A.; Stauber, M. Context-Dependent Functional Divergence of the Notch Ligands DLL1 and DLL4 In Vivo. PLoS Genet. 2015, 11, e1005328. [Google Scholar] [CrossRef]
- Vanhorenbeeck, V.; Jenny, M.; Cornut, J.F.; Gradwohl, G.; Lemaigre, F.P.; Rousseau, G.G.; Jacquemin, P. Role of the Onecut transcription factors in pancreas morphogenesis and in pancreatic and enteric endocrine differentiation. Dev. Biol. 2007, 305, 685–694. [Google Scholar] [CrossRef]
- Wu, F.; Sapkota, D.; Li, R.; Mu, X. Onecut 1 and Onecut 2 are potential regulators of mouse retinal development. J. Comp. Neurol. 2012, 520, 952–969. [Google Scholar] [CrossRef]
- Bochkis, I.M.; Schug, J.; Ye, D.Z.; Kurinna, S.; Stratton, S.A.; Barton, M.C.; Kaestner, K.H. Genome-wide location analysis reveals distinct transcriptional circuitry by paralogous regulators Foxa1 and Foxa2. PLoS Genet. 2012, 8, e1002770. [Google Scholar] [CrossRef]
- Kathiriya, I.S.; Rao, K.S.; Iacono, G.; Devine, W.P.; Blair, A.P.; Hota, S.K.; Lai, M.H.; Garay, B.I.; Thomas, R.; Gong, H.Z.; et al. Modeling Human TBX5 Haploinsufficiency Predicts Regulatory Networks for Congenital Heart Disease. Dev. Cell 2021, 56, 292–309.e299. [Google Scholar] [CrossRef]
- Chowdhury, F.; Wang, L.; Al-Raqad, M.; Amor, D.J.; Baxová, A.; Bendová, Š.; Biamino, E.; Brusco, A.; Caluseriu, O.; Cox, N.J.; et al. Haploinsufficiency of PRR12 causes a spectrum of neurodevelopmental, eye, and multisystem abnormalities. Genet. Med. 2021, 23, 1234–1245. [Google Scholar] [CrossRef]
- Yi, F.; Danko, T.; Botelho, S.C.; Patzke, C.; Pak, C.; Wernig, M.; Südhof, T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016, 352, aaf2669. [Google Scholar] [CrossRef]
- Kummeling, J.; Stremmelaar, D.E.; Raun, N.; Reijnders, M.R.F.; Willemsen, M.H.; Ruiterkamp-Versteeg, M.; Schepens, M.; Man, C.C.O.; Gilissen, C.; Cho, M.T.; et al. Characterization of SETD1A haploinsufficiency in humans and Drosophila defines a novel neurodevelopmental syndrome. Mol. Psychiatry 2021, 26, 2013–2024. [Google Scholar] [CrossRef]
- Zawerton, A.; Mignot, C.; Sigafoos, A.; Blackburn, P.R.; Haseeb, A.; McWalter, K.; Ichikawa, S.; Nava, C.; Keren, B.; Charles, P.; et al. Widening of the genetic and clinical spectrum of Lamb-Shaffer syndrome, a neurodevelopmental disorder due to SOX5 haploinsufficiency. Genet. Med. 2020, 22, 524–537. [Google Scholar] [CrossRef]
- Lalli, M.A.; Jang, J.; Park, J.H.; Wang, Y.; Guzman, E.; Zhou, H.; Audouard, M.; Bridges, D.; Tovar, K.R.; Papuc, S.M.; et al. Haploinsufficiency of BAZ1B contributes to Williams syndrome through transcriptional dysregulation of neurodevelopmental pathways. Hum. Mol. Genet. 2016, 25, 1294–1306. [Google Scholar] [CrossRef]
- Gennarino, V.A.; Palmer, E.E.; McDonell, L.M.; Wang, L.; Adamski, C.J.; Koire, A.; See, L.; Chen, C.A.; Schaaf, C.P.; Rosenfeld, J.A.; et al. A Mild PUM1 Mutation Is Associated with Adult-Onset Ataxia, whereas Haploinsufficiency Causes Developmental Delay and Seizures. Cell 2018, 172, 924–936.e911. [Google Scholar] [CrossRef]
- Hanks, M.; Wurst, W.; Anson-Cartwright, L.; Auerbach, A.B.; Joyner, A.L. Rescue of the En-1 mutant phenotype by replacement of En-1 with En-2. Science 1995, 269, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Chia, I.V.; Costantini, F. Mouse axin and axin2/conductin proteins are functionally equivalent in vivo. Mol. Cell Biol. 2005, 25, 4371–4376. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.J.; Khoriaty, R.; Kiseleva, A.; Cleuren, A.C.A.; Tomberg, K.; van der Ent, M.A.; Gergics, P.; Tang, V.T.; Zhu, G.; Hoenerhoff, M.J.; et al. Murine SEC24D can substitute functionally for SEC24C during embryonic development. Sci. Rep. 2021, 11, 21100. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.; Pfeffer, P.; Busslinger, M. Functional equivalence of the transcription factors Pax2 and Pax5 in mouse development. Development 2000, 127, 3703–3713. [Google Scholar] [CrossRef] [PubMed]
- Savory, J.G.; Pilon, N.; Grainger, S.; Sylvestre, J.R.; Béland, M.; Houle, M.; Oh, K.; Lohnes, D. Cdx1 and Cdx2 are functionally equivalent in vertebral patterning. Dev. Biol. 2009, 330, 114–122. [Google Scholar] [CrossRef]
- Suda, Y.; Nakabayashi, J.; Matsuo, I.; Aizawa, S. Functional equivalency between Otx2 and Otx1 in development of the rostral head. Development 1999, 126, 743–757. [Google Scholar] [CrossRef]
- Acampora, D.; Avantaggiato, V.; Tuorto, F.; Barone, P.; Perera, M.; Choo, D.; Wu, D.; Corte, G.; Simeone, A. Differential transcriptional control as the major molecular event in generating Otx1-/- and Otx2-/- divergent phenotypes. Development 1999, 126, 1417–1426. [Google Scholar] [CrossRef]
- Acampora, D.; Annino, A.; Puelles, E.; Alfano, I.; Tuorto, F.; Simeone, A. OTX1 compensates for OTX2 requirement in regionalisation of anterior neuroectoderm. Gene Expr. Patterns 2003, 3, 497–501. [Google Scholar] [CrossRef]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004, 18, 1088–1105. [Google Scholar] [CrossRef]
- Yang, L.T.; Kaartinen, V. Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev. Biol. 2007, 312, 384–395. [Google Scholar] [CrossRef]
- Carbe, C.; Garg, A.; Cai, Z.; Li, H.; Powers, A.; Zhang, X. An allelic series at the paired box gene 6 (Pax6) locus reveals the functional specificity of Pax genes. J. Biol. Chem. 2013, 288, 12130–12141. [Google Scholar] [CrossRef]
- Wolf, L.; Harrison, W.; Huang, J.; Xie, Q.; Xiao, N.; Sun, J.; Kong, L.; Lachke, S.A.; Kuracha, M.R.; Govindarajan, V.; et al. Histone posttranslational modifications and cell fate determination: Lens induction requires the lysine acetyltransferases CBP and p300. Nucleic Acids Res. 2013, 41, 10199–10214. [Google Scholar] [CrossRef]
- Rocha, S.F.; Lopes, S.S.; Gossler, A.; Henrique, D. Dll1 and Dll4 function sequentially in the retina and pV2 domain of the spinal cord to regulate neurogenesis and create cell diversity. Dev. Biol. 2009, 328, 54–65. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, T.; Madakashira, B.P.; Thiels, C.A.; Bechtle, C.A.; Garcia, C.M.; Zhang, H.; Yu, K.; Ornitz, D.M.; Beebe, D.C.; et al. Fibroblast growth factor receptor signaling is essential for lens fiber cell differentiation. Dev. Biol. 2008, 318, 276–288. [Google Scholar] [CrossRef]
- Munroe, R.J.; Prabhu, V.; Acland, G.M.; Johnson, K.R.; Harris, B.S.; O’Brien, T.P.; Welsh, I.C.; Noden, D.M.; Schimenti, J.C. Mouse H6 Homeobox 1 (Hmx1) mutations cause cranial abnormalities and reduced body mass. BMC Dev. Biol. 2009, 9, 27. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Nichini, O.; Boisset, G.; Polok, B.; Tiab, L.; Mayeur, H.; Raji, B.; de la Houssaye, G.; Abitbol, M.M.; Munier, F.L. Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome. Am. J. Hum. Genet. 2008, 82, 1178–1184. [Google Scholar] [CrossRef]
- Fujimoto, M.; Izu, H.; Seki, K.; Fukuda, K.; Nishida, T.; Yamada, S.; Kato, K.; Yonemura, S.; Inouye, S.; Nakai, A. HSF4 is required for normal cell growth and differentiation during mouse lens development. Embo J. 2004, 23, 4297–4306. [Google Scholar] [CrossRef]
- Somasundaram, T.; Bhat, S.P. Developmentally dictated expression of heat shock factors: Exclusive expression of HSF4 in the postnatal lens and its specific interaction with alphaB-crystallin heat shock promoter. J. Biol. Chem. 2004, 279, 44497–44503. [Google Scholar] [CrossRef]
- Antosova, B.; Smolikova, J.; Klimova, L.; Lachova, J.; Bendova, M.; Kozmikova, I.; Machon, O.; Kozmik, Z. The Gene Regulatory Network of Lens Induction Is Wired through Meis-Dependent Shadow Enhancers of Pax6. PLoS Genet. 2016, 12, e1006441. [Google Scholar] [CrossRef]
- Dupacova, N.; Antosova, B.; Paces, J.; Kozmik, Z. Meis homeobox genes control progenitor competence in the retina. Proc. Natl. Acad. Sci. USA 2021, 118, e2013136118. [Google Scholar] [CrossRef]
- Kreplova, M.; Kuzelova, A.; Antosova, B.; Zilova, L.; Jägle, H.; Kozmik, Z. Dose-dependent regulation of horizontal cell fate by Onecut family of transcription factors. PLoS ONE 2020, 15, e0237403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, W.-T.; Du, W.; Zhu, P.; Li, J.; Xu, F.; Sun, J.; Gerstner, C.D.; Baehr, W.; Boye, S.L.; et al. Gene-based Therapy in a Mouse Model of Blue Cone Monochromacy. Sci. Rep. 2017, 7, 6690. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Johnson, S.; Simunovic, M.P.; Bradshaw, K.; Holder, G.; Mollon, J.D.; Moore, A.T.; Hunt, D.M. Blue cone monochromatism: A phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye 2005, 19, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Neitz, J.; Neitz, M. The genetics of normal and defective color vision. Vis. Res. 2011, 51, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Bosze, B.; Suarez-Navarro, J.; Soofi, A.; Lauderdale, J.D.; Dressler, G.R.; Brown, N.L. Multiple roles for Pax2 in the embryonic mouse eye. Dev. Biol. 2021, 472, 18–29. [Google Scholar] [CrossRef]
- Diacou, R.; Zhao, Y.; Zheng, D.; Cvekl, A.; Liu, W. Six3 and Six6 Are Jointly Required for the Maintenance of Multipotent Retinal Progenitors through Both Positive and Negative Regulation. Cell Rep. 2018, 25, 2510–2523.e2514. [Google Scholar] [CrossRef]
- Fromental-Ramain, C.; Warot, X.; Lakkaraju, S.; Favier, B.; Haack, H.; Birling, C.; Dierich, A.; Doll e, P.; Chambon, P. Specific and redundant functions of the paralogous Hoxa-9 and Hoxd-9 genes in forelimb and axial skeleton patterning. Development 1996, 122, 461–472. [Google Scholar] [CrossRef]
- Gerner-Mauro, K.N.; Akiyama, H.; Chen, J. Redundant and additive functions of the four Lef/Tcf transcription factors in lung epithelial progenitors. Proc. Natl. Acad. Sci. USA 2020, 117, 12182–12191. [Google Scholar] [CrossRef]
- Hagelkruys, A.; Lagger, S.; Krahmer, J.; Leopoldi, A.; Artaker, M.; Pusch, O.; Zezula, J.; Weissmann, S.; Xie, Y.; Schöfer, C.; et al. A single allele of Hdac2 but not Hdac1 is sufficient for normal mouse brain development in the absence of its paralog. Development 2014, 141, 604–616. [Google Scholar] [CrossRef]
- Böhm, J.; Buck, A.; Borozdin, W.; Mannan, A.U.; Matysiak-Scholze, U.; Adham, I.; Schulz-Schaeffer, W.; Floss, T.; Wurst, W.; Kohlhase, J.; et al. Sall1, sall2, and sall4 are required for neural tube closure in mice. Am. J. Pathol. 2008, 173, 1455–1463. [Google Scholar] [CrossRef]
- Schmitz-Rohmer, D.; Probst, S.; Yang, Z.Z.; Laurent, F.; Stadler, M.B.; Zuniga, A.; Zeller, R.; Hynx, D.; Hemmings, B.A.; Hergovich, A. NDR Kinases Are Essential for Somitogenesis and Cardiac Looping during Mouse Embryonic Development. PLoS ONE 2015, 10, e0136566. [Google Scholar] [CrossRef]
- Carlisle, F.A.; Pearson, S.; Steel, K.P.; Lewis, M.A. Pitpnm1 is expressed in hair cells during development but is not required for hearing. Neuroscience 2013, 248, 620–625. [Google Scholar] [CrossRef]
- Jin, J.Z.; Ding, J. Strain-dependent effects of transforming growth factor-β1 and 2 during mouse secondary palate development. Reprod Toxicol. 2014, 50, 129–133. [Google Scholar] [CrossRef]
- Gómez-Redondo, I.; Ramos-Ibeas, P.; Pericuesta, E.; Fernández-González, R.; Laguna-Barraza, R.; Gutiérrez-Adán, A. Minor Splicing Factors Zrsr1 and Zrsr2 Are Essential for Early Embryo Development and 2-Cell-Like Conversion. Int. J. Mol. Sci. 2020, 21, 14115. [Google Scholar] [CrossRef]
- Wang, G.; Ying, Z.; Jin, X.; Tu, N.; Zhang, Y.; Phillips, M.; Moskophidis, D.; Mivechi, N.F. Essential requirement for both hsf1 and hsf2 transcriptional activity in spermatogenesis and male fertility. Genesis 2004, 38, 66–80. [Google Scholar] [CrossRef]
- Collette, N.M.; Yee, C.S.; Murugesh, D.; Sebastian, A.; Taher, L.; Gale, N.W.; Economides, A.N.; Harland, R.M.; Loots, G.G. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev. Biol. 2013, 383, 90–105. [Google Scholar] [CrossRef]
- Gao, Y.; Lan, Y.; Liu, H.; Jiang, R. The zinc finger transcription factors Osr1 and Osr2 control synovial joint formation. Dev. Biol. 2011, 352, 83–91. [Google Scholar] [CrossRef]
- Yoshida, T.; Miyoshi, J.; Takai, Y.; Thesleff, I. Cooperation of nectin-1 and nectin-3 is required for normal ameloblast function and crown shape development in mouse teeth. Dev. Dyn. 2010, 239, 2558–2569. [Google Scholar] [CrossRef]
- Batrakou, D.G.; de Las Heras, J.I.; Czapiewski, R.; Mouras, R.; Schirmer, E.C. TMEM120A and B: Nuclear Envelope Transmembrane Proteins Important for Adipocyte Differentiation. PLoS ONE 2015, 10, e0127712. [Google Scholar] [CrossRef]
- Boucherat, O.; Montaron, S.; Bérubé-Simard, F.A.; Aubin, J.; Philippidou, P.; Wellik, D.M.; Dasen, J.S.; Jeannotte, L. Partial functional redundancy between Hoxa5 and Hoxb5 paralog genes during lung morphogenesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L817–L830. [Google Scholar] [CrossRef]
- Larsen, B.M.; Hrycaj, S.M.; Newman, M.; Li, Y.; Wellik, D.M. Mesenchymal Hox6 function is required for mouse pancreatic endocrine cell differentiation. Development 2015, 142, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gerber, A.; Chen, W.Y.; Roeder, R.G. Functions of paralogous RNA polymerase III subunits POLR3G and POLR3GL in mouse development. Proc. Natl. Acad. Sci. USA 2020, 117, 15702–15711. [Google Scholar] [CrossRef] [PubMed]
- Bendall, A.J. Direct evidence of allele equivalency at the Dlx5/6 locus. Genesis 2016, 54, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Yallowitz, A.R.; Hrycaj, S.M.; Short, K.M.; Smyth, I.M.; Wellik, D.M. Hox10 genes function in kidney development in the differentiation and integration of the cortical stroma. PLoS ONE 2011, 6, e23410. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashida, N.; Katoh, T.; Oshima, K.; Shinkawa, T.; Prakasam, R.; Tan, K.; Inouye, S.; Takii, R.; Nakai, A. A novel mouse HSF3 has the potential to activate nonclassical heat-shock genes during heat shock. Mol. Biol. Cell 2010, 21, 106–116. [Google Scholar] [CrossRef]
- Peters, H.; Wilm, B.; Sakai, N.; Imai, K.; Maas, R.; Balling, R. Pax1 and Pax9 synergistically regulate vertebral column development. Development 1999, 126, 5399–5408. [Google Scholar] [CrossRef]
- Manley, N.R.; Capecchi, M.R. HOX group 3 paralogous genes act synergistically in the formation of somitic and neural crest-derived structures. Dev. Biol. 1997, 192, 274–288. [Google Scholar] [CrossRef]
- Boulet, A.M.; Capecchi, M.R. Targeted disruption of hoxc-4 causes esophageal defects and vertebral transformations. Dev. Biol. 1996, 177, 232–249. [Google Scholar] [CrossRef]
- Wahba, G.M.; Hostikka, S.L.; Carpenter, E.M. The paralogous HOX genes Hoxa10 and Hoxd10 interact to pattern the mouse hindlimb peripheral nervous system and skeleton. Dev. Biol. 2001, 231, 87–102. [Google Scholar] [CrossRef]
- Ashokkumar, D.; Zhang, Q.; Much, C.; Bledau, A.S.; Naumann, R.; Alexopoulou, D.; Dahl, A.; Goveas, N.; Fu, J.; Anastassiadis, K.; et al. MLL4 is required after implantation, whereas MLL3 becomes essential during late gestation. Development 2020, 147, dev186999. [Google Scholar] [CrossRef]
- Kobayashi, K.; Endo, T.; Matsumura, T.; Lu, Y.; Yu, Z.; Matzuk, M.M.; Ikawa, M. Prss55 but not Prss51 is required for male fertility in mice. Biol. Reprod. 2020, 103, 223–234. [Google Scholar] [CrossRef]
- Devlin, D.J.; Nozawa, K.; Ikawa, M.; Matzuk, M.M. Knockout of family with sequence similarity 170 member A (Fam170a) causes male subfertility, while Fam170b is dispensable in mice†. Biol. Reprod. 2020, 103, 205–222. [Google Scholar] [CrossRef]
- Bledau, A.S.; Schmidt, K.; Neumann, K.; Hill, U.; Ciotta, G.; Gupta, A.; Torres, D.C.; Fu, J.; Kranz, A.; Stewart, A.F.; et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 2014, 141, 1022–1035. [Google Scholar] [CrossRef]
- Bahat, A.; Kedmi, R.; Gazit, K.; Richardo-Lax, I.; Ainbinder, E.; Dikstein, R. TAF4b and TAF4 differentially regulate mouse embryonic stem cells maintenance and proliferation. Genes Cells 2013, 18, 225–237. [Google Scholar] [CrossRef]
- Messiaen, S.; Guiard, J.; Aigueperse, C.; Fliniaux, I.; Tourpin, S.; Barroca, V.; Allemand, I.; Fouchet, P.; Livera, G.; Vernet, M. Loss of the histone chaperone ASF1B reduces female reproductive capacity in mice. Reproduction 2016, 151, 477–489. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Dou, J.; Yu, T.; Cao, P.; Fan, N.; Borjigin, U.; Nashun, B. Distinct role of histone chaperone Asf1a and Asf1b during fertilization and pre-implantation embryonic development in mice. Epigenetics Chromatin 2021, 14, 55. [Google Scholar] [CrossRef]
- Akbas, G.E.; Taylor, H.S. HOXC and HOXD gene expression in human endometrium: Lack of redundancy with HOXA paralogs. Biol. Reprod. 2004, 70, 39–45. [Google Scholar] [CrossRef]
- Winderickx, J.; Battisti, L.; Motulsky, A.G.; Deeb, S.S. Selective expression of human X chromosome-linked green opsin genes. Proc. Natl. Acad. Sci. USA 1992, 89, 9710–9714. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Motulsky, A.G.; Deeb, S.S. Visual Pigment Gene Structure and Expression in Human Retinae. Hum. Mol. Genet. 1997, 6, 981–990. [Google Scholar] [CrossRef]
- Applebury, M.L.; Antoch, M.P.; Baxter, L.C.; Chun, L.L.Y.; Falk, J.D.; Farhangfar, F.; Kage, K.; Krzystolik, M.G.; Lyass, L.A.; Robbins, J.T. The Murine Cone Photoreceptor: A Single Cone Type Expresses Both S and M Opsins with Retinal Spatial Patterning. Neuron 2000, 27, 513–523. [Google Scholar] [CrossRef]
- Stamboulian, M.; Guerrero, R.F.; Hahn, M.W.; Radivojac, P. The ortholog conjecture revisited: The value of orthologs and paralogs in function prediction. Bioinformatics 2020, 36, i219–i226. [Google Scholar] [CrossRef] [PubMed]
- Dandage, R.; Landry, C.R. Paralog dependency indirectly affects the robustness of human cells. Mol. Syst. Biol. 2019, 15, e8871. [Google Scholar] [CrossRef] [PubMed]
- Diss, G.; Ascencio, D.; DeLuna, A.; Landry, C.R. Molecular mechanisms of paralogous compensation and the robustness of cellular networks. J. Exp. Zool B Mol. Dev. Evol. 2014, 322, 488–499. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).