
Complete Mitogenome of Oreolalax omeimontis Reveals Phylogenetic Status and Novel Gene Arrangement of Archaeobatrachia

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. PCR Amplification and Sequencing
2.3. Sequence Assembly, Annotation, and Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
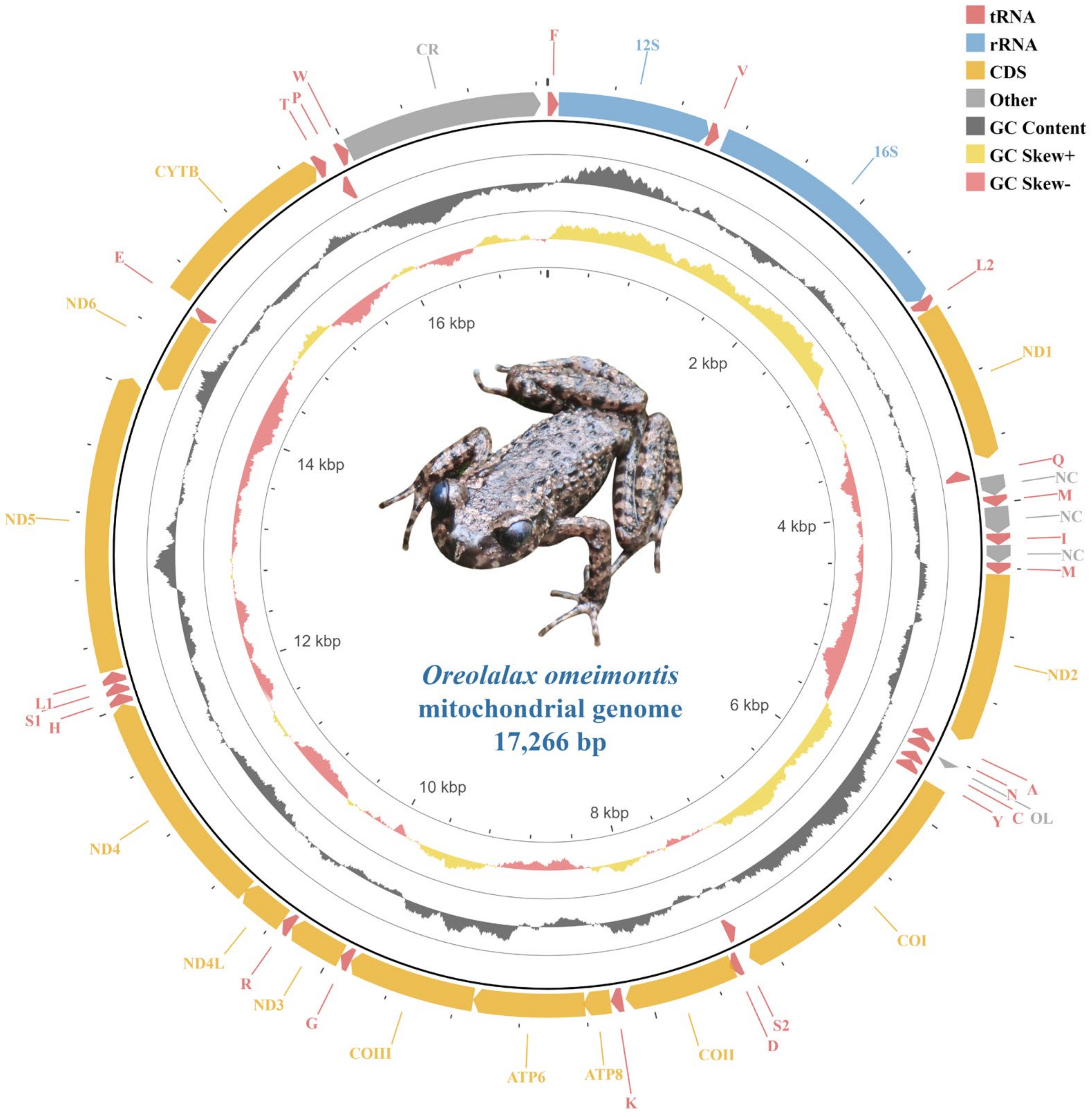
3.1. Mitogenome Organization
3.2. Protein-Coding Genes and Codon Usage
3.3. Transfer RNA
3.4. Ribosomal RNA and Control Region
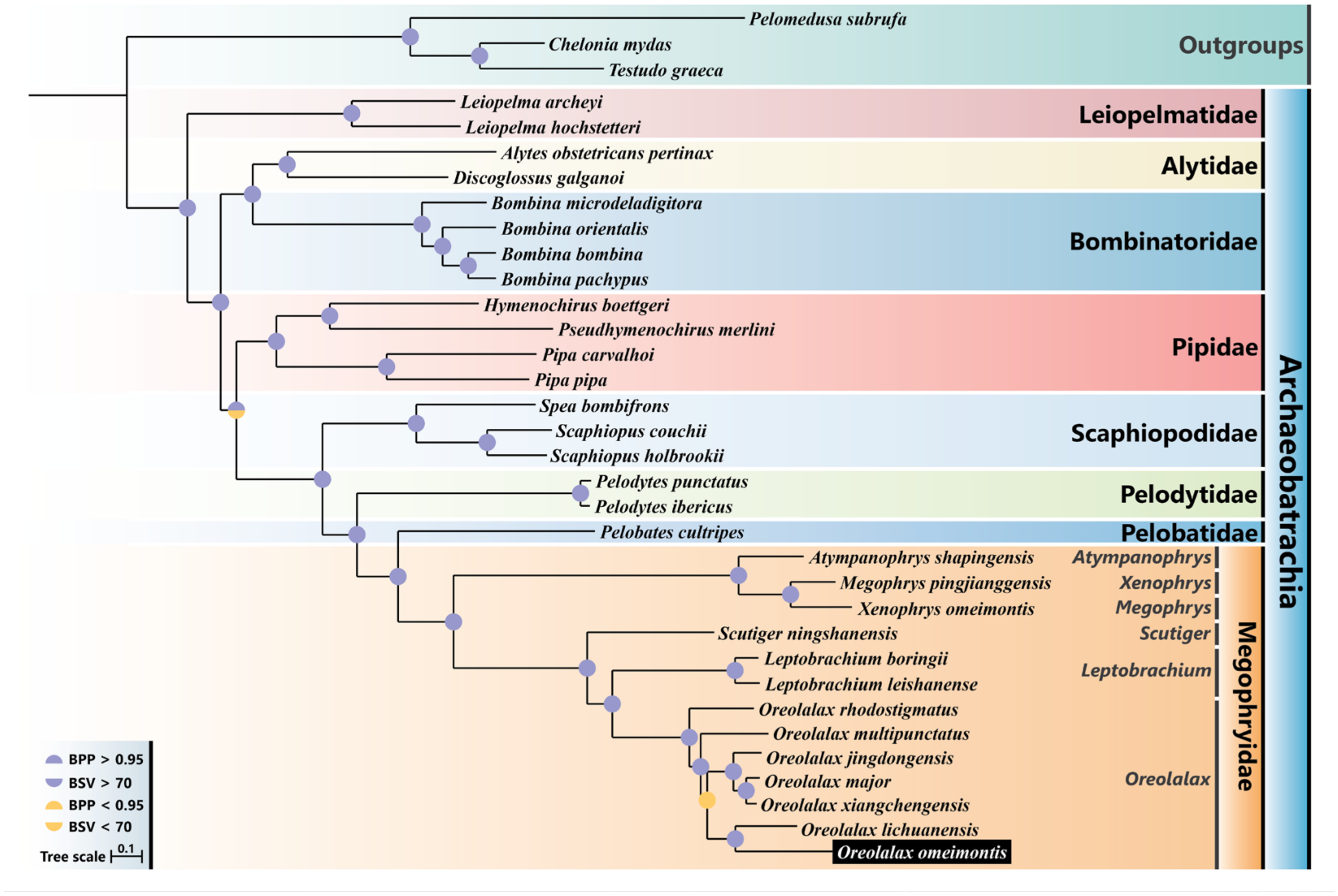
3.5. Phylogenetic Analysis
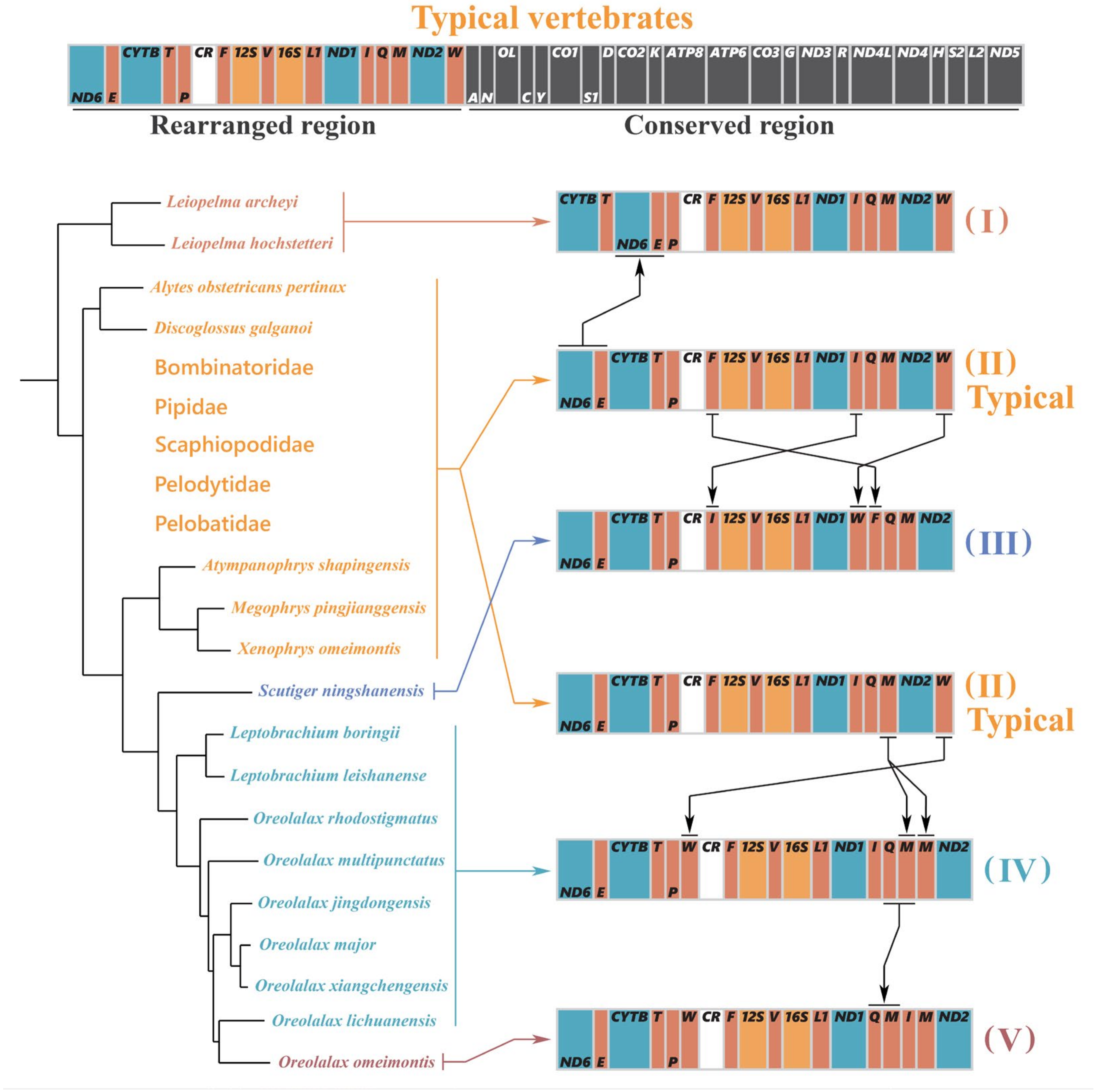
3.6. Gene Rearrangement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Podsiadlowski, L.; Kohlhagen, H.; Koch, M. The complete mitochondrial genome of Scutigerella causeyae (Myriapoda: Symphyla) and the phylogenetic position of Symphyla. Mol. Phylogenetics Evol. 2007, 45, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Unmack, P.J.; Kuchling, G.; Wang, Y.; Georges, A. Resolution of the enigmatic phylogenetic relationship of the critically endangered Western Swamp Tortoise Pseudemydura umbrina (Pleurodira: Chelidae) using a complete mitochondrial genome. Mol. Phylogenet. Evol. 2017, 115, 58–61. [Google Scholar] [CrossRef]
- Fei, L.; Shushen, L.I. On the Generic Calssification of Asian High Altitude Pelobatid Toads (Amphibia: Pelobatidae). Acta Zool. Sin. 1989, 35, 381–389. [Google Scholar]
- Mcdonough, K. Amphibian Species of the World: An Online Reference (Version 6). Ref. Rev. 2014, 28, 32. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Phung, T.M.; Le, M.D.; Ziegler, T.; Böhme, W. First record of the genus Oreolalax (Anura: Megophryidae) from Vietnam with description of a new species. Copeia 2013, 2013, 213–222. [Google Scholar] [CrossRef]
- Wei, G.; Wang, B.; Xu, N.; Li, Z.; Jiang, J. Morphological evolution from aquatic to terrestrial in the genus Oreolalax (Amphibia, Anura, Megophryidae). Prog. Nat. Sci. 2009, 19, 1403–1408. [Google Scholar] [CrossRef]
- Wiens, J.J.; Sukumaran, J.; Pyron, R.A.; Brown, R.M. Evolutionary and biogeographic origins of high tropical diversity in old world frogs (Ranidae). Evolution 2009, 63, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.X.; Wang, B.; Li, C.; Xiang, T.M.; Jiang, J.P.; Xie, F. The complete mitochondrial genome of Oreolalax major (Anura: Megophryidae). Mitochondrial Dna Part B Resour. 2016, 1, 118–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Li, S.; Le, W.; Gao, X.; Gang, W.; Wang, B. The complete mitochondrial genome of the toad species Oreolalax jingdongensis (Anura: Megophryidae) and related phylogenetic analyses. Conserv. Genet. Resour. 2017, 10, 873–876. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, J.; Zhe, W.; Zhang, Z.; Wu, M.; Lei, Y. The complete mitochondrial genome of the vulnerable megophryid frog Oreolalax rhodostigmatus (Anura, Megophryidae). Conserv. Genet. Resour. 2017, 10, 617–620. [Google Scholar] [CrossRef]
- Li, S.; Gao, X.; Jiang, J.; Wang, B. Complete mitogenome of the toad species Oreolalax multipunctatus (Anura: Megophryidae) and phylogenetic analyses of Megophryidae. Conserv. Genet. Resour. 2018, 11, 195–198. [Google Scholar] [CrossRef]
- Li, S.; Gao, X.; Wei, G.; Wang, B.; Xu, N. The complete mitochondrial genome of the toad species Oreolalax xiangchengensis (Anura: Megophryidae) and phylogenetic analyses. Mitochondrial DNA Part B 2018, 3, 1298–1299. [Google Scholar] [CrossRef] [Green Version]
- Roe, B.A.; Ma, D.P.; Wilson, R.K.; Wong, J.F. The complete nucleotide sequence of the Xenopus laevis mitochondrial genome. J. Biol. Chem. 1985, 260, 9759. [Google Scholar] [CrossRef]
- Pabijan, M.; Spolsky, C.; Uzzell, T.; Szymura, J.M. Comparative Analysis of Mitochondrial Genomes in Bombina (Anura; Bombinatoridae). J. Mol. Evol. 2008, 67, 246–256. [Google Scholar] [CrossRef]
- Xia, Y.; Zheng, Y.; Miura, I.; Wong, P.B.; Murphy, R.W.; Zeng, X. The evolution of mitochondrial genomes in modern frogs (Neobatrachia): Nonadaptive evolution of mitochondrial genome reorganization. BMC Genom. 2014, 15, 691. [Google Scholar] [CrossRef] [Green Version]
- Fei, L.; Ye, C.; Huang, Y.; Liu, M. Atlas of Amphibians of China; Henan Science and Technology Press: Zhengzhou, China, 1999. [Google Scholar]
- Frank, N.; Ramus, E. A Complete Guide to Scientific and Common Names of Reptiles and Amphibians of the World; NG Publishing, Inc.: Pottsville, PA, USA, 1995. [Google Scholar]
- AmphibiaChina. The Database of Chinese Amphibians. Available online: http://www.amphibiachina.org/ (accessed on 10 May 2020).
- Fu, J.-Z.; Weadick, C.; Bi, K. A phylogeny of the high-elevation Tibetan megophryid frogs and evidence for the multiple origins of reversed sexual size dimorphism. J. Zool. 2007, 273, 315–325. [Google Scholar] [CrossRef]
- Pyron, R.A.; Wiens, J.J. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylogenet. Evol. 2011, 61, 543–583. [Google Scholar] [CrossRef] [PubMed]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, T.A.; Macinnis, M.J.; Bieler, R.; Boore, J.L.; Collins, T.M. Sessile snails, dynamic genomes: Gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genom. 2010, 11, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, A.; Golombek, A.; Gerth, M.; Schwarz, F.; Struck, T.H.; Bleidorn, C. Evolution of mitochondrial gene order in Annelida. Mol. Phylogenet. Evol. 2016, 94, 196–206. [Google Scholar] [CrossRef]
- Boore, J.L. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol. Evol. 2006, 21, 439–446. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, J.; Li, B.; Ouyang, B.; Yang, J. The complete mitochondrial genome of Budorcas taxicolor tibetana (Artiodactyla: Bovidae) and comparison with other Caprinae species: Insight into the phylogeny of the genus Budorcas. Int. J. Biol. Macromol. 2019, 121, 223–232. [Google Scholar] [CrossRef]
- Zhang, P.; Liang, D.; Mao, R.-L.; Hillis, D.M.; Wake, D.B.; Cannatella, D.C. Efficient Sequencing of Anuran mtDNAs and a Mitogenomic Exploration of the Phylogeny and Evolution of Frogs. Mol. Biol. Evol. 2013, 30, 1899–1915. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Peter, S.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar]
- Grant, J.R.; Paul, S. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Kazutaka, K.; Kazuharu, M.; Kei-Ichi, K.; Takashi, M. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Haeseler, A.V.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Li, W.; Jakovlić, I. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. bioRxiv 2018. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, gkw290. [Google Scholar] [CrossRef]
- Fearnley, I.M.; Walker, J.E. Initiation codons in mammalian mitochondria: Differences in genetic code in the organelle. Biochemistry 1987, 26, 8247. [Google Scholar] [CrossRef]
- Satoh, T.P.; Al, E. Round and pointed-head grenadier fishes (Actinopterygii: Gadiformes) represent a single sister group: Evidence from the complete mitochondrial genome sequences. Mol. Phylogenet. Evol. 2006, 40, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichert, A.S.; Mörl, M. Repair of tRNAs in metazoan mitochondria. Nucleic Acids Res. 2000, 28, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, R.; Wolstenholme, D.R. A set of tRNAs that lack either the T psi C arm or the dihydrouridine arm: Towards a minimal tRNA adaptor. EMBO J. 1990, 9, 3405–3411. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.J.; Blackburn, D.C.; Liang, D.; Hillis, D.M.; Wake, D.B.; Cannatella, D.C.; Zhang, P. Phylogenomics reveals rapid, simultaneous diversification of three major clades of Gondwanan frogs at the Cretaceous-Paleogene boundary. Proc. Natl. Acad. Sci. USA 2017, 114, 201704632. [Google Scholar] [CrossRef] [Green Version]
- Kurabayashi, A.; Sumida, M.; Yonekawa, H.; Glaw, F.; Vences, M.; Hasegawa, M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 2008, 25, 874–891. [Google Scholar] [CrossRef] [Green Version]
- Mark, D.; Cameron, S.L.; Dowavic, J.I.; Austin, A.D.; Whiting, M.F. Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol. Biol. Evol. 2009, 26, 1607–1617. [Google Scholar]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Sumida, M.; Kanamori, Y.H.; Kato, Y.; Nishioka, M.; Hasegawa, M. Complete nucleotide sequence and gene rearrangement of the mitochondria genome of the Japanese pond frog Rana nigromaculata. Genes Genet. Syst. 2001, 76, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Mohammad Shafiqul, A.; Atsushi, K.; Yoko, H.; Naomi, S.; Mukhlesur Rahman, K.; Tamotsu, F.; Masayuki, S. Complete mitochondrial genomes and novel gene rearrangements in two dicroglossid frogs, Hoplobatrachus tigerinus and Euphlyctis hexadactylus, from Bangladesh. Genes Genet. Syst. 2010, 85, 219. [Google Scholar]
- Carr, L.M.; Mclenachan, T.; Waddell, P.J.; Gemmell, N.J.; Penny, D. Analyses of the mitochondrial genome of Leiopelma hochstetteri argues against the full drowning of New Zealand. J. Biogeogr. 2015, 42, 1066–1076. [Google Scholar] [CrossRef]
- Song, J.; Tian, Y.; Guan, D.L. Characterization of the complete mitochondrial genome of an endangered alpine toad, Scutiger ningshanensis (Amphibia: Anura: Megophryidae). Conserv. Genet. Resour. 2016, 9, 35–38. [Google Scholar] [CrossRef]
- Macey, J.R.; Larson, A.; Ananjeva, N.B.; Fang, Z.; Papenfuss, T.J. Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol. Biol. Evol. 2007, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Okajima, Y.; Kumazawa, Y. Mitochondrial genomes of acrodont lizards: Timing of gene rearrangements and phylogenetic and biogeographic implications. BMC Evol. Biol. 2010, 10, 141. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Strand | Location | Size (bp) | Anti-codon | Start Codon | Stop Codon | IGS 1 |
|---|---|---|---|---|---|---|---|
| trnF | + | 1–68 | 68 | GAA | 0 | ||
| rrnS | + | 69–1003 | 935 | 0 | |||
| trnV | + | 1004–1072 | 69 | TAC | 0 | ||
| rrnL | + | 1073-2672 | 1600 | 0 | |||
| trnL2 | + | 2673–2747 | 75 | TAA | 0 | ||
| nad1 | + | 2748–3725 | 978 | ATG | TAA | 38 | |
| trnQ | – | 3764–3834 | 71 | TTG | 21 | ||
| trnM | + | 3956–4023 | 68 | CAT | 163 | ||
| trnI | + | 4187–4258 | 72 | GAT | 107 | ||
| trnM | + | 4366–4435 | 72 | CAT | 1 | ||
| nad2 | + | 4437–5480 | 1044 | ATG | TAA | 10 | |
| trnA | – | 5491–5560 | 70 | TGC | 0 | ||
| trnN | – | 5561–5633 | 73 | GTT | 2 | ||
| OL | + | 5636–5662 | 27 | −1 | |||
| trnC | – | 5662–5725 | 64 | GCA | 0 | ||
| trnY | – | 5726–5795 | 70 | GTA | 1 | ||
| cox1 | + | 5797–7359 | 1563 | GTG | AGG | −9 | |
| trnS2 | – | 7351–7421 | 71 | TGA | 4 | ||
| trnD | + | 7426–7493 | 68 | GTC | 0 | ||
| cox2 | + | 7494-8175 | 682 | ATG | T(AA) | 0 | |
| trnK | + | 8176–8249 | 74 | TTT | 0 | ||
| atp8 | + | 8250–8417 | 168 | ATG | TAA | −10 | |
| atp6 | + | 8408–9090 | 683 | ATG | TA(A) | −1 | |
| cox3 | + | 9090–9874 | 785 | ATG | TA(A) | −1 | |
| trnG | + | 9874–9943 | 70 | TCC | 0 | ||
| nad3 | + | 9944-10,286 | 343 | ATG | T(AA) | 0 | |
| trnR | + | 10,287–10,355 | 69 | TCG | 0 | ||
| nad4l | + | 10,356–10,652 | 297 | ATG | TAA | −7 | |
| nad4 | + | 10,646–12,024 | 1379 | ATG | TA(A) | −1 | |
| trnH | + | 12,024–12,092 | 69 | GTG | 0 | ||
| trnS1 | + | 12,093–12,159 | 67 | GCT | 0 | ||
| trnL1 | + | 12,160–12,231 | 72 | TAG | 6 | ||
| nad5 | + | 12,238–14,058 | 1821 | ATT | AGG | 4 | |
| nad6 | – | 14,063–14,572 | 510 | GTG | AGG | 0 | |
| trnE | – | 14,573–14,641 | 69 | TTC | 3 | ||
| cob | + | 14,645–15,785 | 1141 | ATG | T(AA) | 0 | |
| trnT | + | 15,786–15,855 | 70 | TGT | 2 | ||
| trnP | – | 15,858–15,926 | 69 | TGG | 15 | ||
| trnW | – | 15,942–16,010 | 69 | TCA | 0 | ||
| CR | + | 16,011–17,266 | 1166 | 0 |
| Species | Complete Mitogenome | tRNAs | PCGs | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A + T% | AT-Skew | GC-Skew | A + T% | AT-Skew | GC-Skew | A + T% | AT-Skew | GC-Skew | |
| O. omeimontis | 58.9 | −0.063 | −0.278 | 58.1 | 0.026 | 0.031 | 58.5 | −0.145 | −0.282 |
| O. lichuanensis | 60.2 | −0.070 | −0.248 | 58.5 | 0.022 | 0.055 | 59.8 | −0.151 | −0.249 |
| O. jingdongensis | 61.8 | −0.058 | −0.251 | 57.9 | 0.036 | 0.040 | 61.5 | −0.138 | −0.252 |
| O. xiangchengensis | 62.2 | −0.061 | −0.249 | 58.4 | 0.027 | 0.017 | 62.5 | −0.142 | −0.248 |
| O. rhodostigmatus | 60.4 | −0.073 | −0.258 | 57.8 | 0.017 | 0.047 | 59.8 | −0.140 | −0.254 |
| O. multipunctatus | 61.5 | −0.073 | −0.257 | 59.3 | 0.025 | 0.039 | 60.8 | −0.145 | −0.250 |
| O. major | 61.2 | −0.059 | −0.260 | 57.4 | 0.035 | 0.047 | 61.3 | −0.132 | −0.266 |
| Species | rRNAs | CR | |||||||
| A + T% | AT-Skew | GC-Skew | A + T% | AT-Skew | GC-Skew | ||||
| O. omeimontis | 58.2 | 0.124 | −0.105 | 63.9 | −0.095 | −0.291 | |||
| O. lichuanensis | 59.5 | 0.109 | −0.101 | 62.6 | −0.089 | −0.212 | |||
| O. jingdongensis | 59.9 | 0.129 | −0.107 | 67.0 | −0.084 | −0.261 | |||
| O. xiangchengensis | 60.3 | 0.121 | −0.106 | 63.7 | −0.068 | −0.247 | |||
| O. rhodostigmatus | 59.3 | 0.116 | −0.115 | 65.4 | −0.125 | −0.246 | |||
| O. multipunctatus | 60.0 | 0.107 | −0.110 | 68.4 | −0.099 | −0.325 | |||
| O. major | 59.5 | 0.126 | −0.104 | 63.9 | −0.121 | −0.247 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, H.; Cui, L.; Han, F.; He, Z.; Fan, X.; Zeng, B.; Yang, M.; Yang, D.; Ni, Q.; Li, Y.; et al. Complete Mitogenome of Oreolalax omeimontis Reveals Phylogenetic Status and Novel Gene Arrangement of Archaeobatrachia. Genes 2022, 13, 2089. https://doi.org/10.3390/genes13112089
Luo H, Cui L, Han F, He Z, Fan X, Zeng B, Yang M, Yang D, Ni Q, Li Y, et al. Complete Mitogenome of Oreolalax omeimontis Reveals Phylogenetic Status and Novel Gene Arrangement of Archaeobatrachia. Genes. 2022; 13(11):2089. https://doi.org/10.3390/genes13112089
Chicago/Turabian StyleLuo, Hongdi, Lin Cui, Fuyao Han, Zhi He, Xiaolan Fan, Bo Zeng, Mingyao Yang, Deying Yang, Qingyong Ni, Yan Li, and et al. 2022. "Complete Mitogenome of Oreolalax omeimontis Reveals Phylogenetic Status and Novel Gene Arrangement of Archaeobatrachia" Genes 13, no. 11: 2089. https://doi.org/10.3390/genes13112089
APA StyleLuo, H., Cui, L., Han, F., He, Z., Fan, X., Zeng, B., Yang, M., Yang, D., Ni, Q., Li, Y., Yao, Y., Xu, H., Yang, J., Wei, Z., Li, T., Rao, D., Yan, T., & Zhang, M. (2022). Complete Mitogenome of Oreolalax omeimontis Reveals Phylogenetic Status and Novel Gene Arrangement of Archaeobatrachia. Genes, 13(11), 2089. https://doi.org/10.3390/genes13112089