
Microsatellite Characterization and Panel Selection for Brown Bear (Ursus arctos) Population Assessment
 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. Microsatellite Genotyping and Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeSalle, R.; Amato, G. The expansion of conservation genetics. Nat. Rev. Genet. 2004, 5, 702–712. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: Retrospect and prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Batter, T.J.; Bush, J.P.; Sacks, B.N. Assessing genetic diversity and connectivity in a tule elk (Cervus canadensis nannodes) metapopulation in Northern California. Conserv. Genet. 2021, 22, 889–901. [Google Scholar] [CrossRef]
- Zilko, J.P.; Harley, D.; Pavlova, A.; Sunnucks, P. Applying population viability analysis to inform genetic rescue that preserves locally unique genetic variation in a critically endangered mammal. Diversity 2021, 13, 382. [Google Scholar] [CrossRef]
- De Barba, M.; Waits, L.P.; Garton, E.O.; Genovesi, P.; Randi, E.; Mustoni, A.; Groff, C. The power of genetic monitoring for studying demography, ecology and genetics of a reintroduced brown bear population. Mol. Ecol. 2010, 19, 3938–3951. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, A.A.; Straka, M.; Drosopoulou, E.; de Gabriel Hernando, M.; Kocijan, I.; Paule, L.; Scouras, Z. Genetic diversity, structure, and size of an endangered brown bear population threatened by highway construction in the Pindos Mountains, Greece. Eur. J. Wildl. Res. 2012, 58, 511–522. [Google Scholar] [CrossRef]
- Haanes, H.; Markussen, S.S.; Herfindal, I.; Røed, K.H.; Solberg, E.J.; Heim, M.; Midthjell, L.; Sæther, B.E. Effects of inbreeding on fitness-related traits in a small isolated moose population. Ecol. Evol. 2013, 3, 4230–4242. [Google Scholar] [CrossRef] [PubMed]
- Brzeski, K.E.; Rabon, D.R., Jr.; Chamberlain, M.J.; Waits, L.P.; Taylor, S.S. Inbreeding and inbreeding depression in endangered red wolves (Canis rufus). Mol. Ecol. 2014, 23, 4241–4255. [Google Scholar] [CrossRef]
- Ekblom, R.; Aronsson, M.; Elsner-Gearing, F.; Johansson, M.; Fountain, T.; Persson, J. Sample identification and pedigree reconstruction in Wolverine (Gulo gulo) using SNP genotyping of non-invasive samples. Conserv. Genet. Resour. 2021, 13, 261–274. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Funk, W.C.; Rajora, O.P. Population genomics for wildlife conservation and management. Mol. Ecol. 2021, 30, 62–82. [Google Scholar] [CrossRef]
- Gervasi, V.; Ciucci, P.; Boulanger, J.; Posillico, M.; Sulli, C.; Focardi, S.; Randi, E.; Boitani, L. A preliminary estimate of the Apennine brown bear population size based on hair-snag sampling and multiple data source mark–recapture Huggins models. Ursus 2008, 19, 105–121. [Google Scholar] [CrossRef]
- Kindberg, J.; Swenson, J.E.; Ericsson, G.; Bellemain, E.; Miquel, C.; Taberlet, P. Estimating population size and trends of the Swedish brown bear Ursus arctos population. Wildl. Biol. 2011, 17, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Tsaparis, D.; Triantafyllidis, A.; Karaiskou, N.; Mertzanis, Y. Νon invasive genetic study and population monitoring of brown bear (Ursus arctos) in the Kastoria region, Greece. In Proceedings of the 11th International Congress on the Zoogeography and Ecology of Greece and Adjacent Regions, Athens, Greece, 18–22 June 2012. [Google Scholar]
- Burgess, B.T.; Irvine, R.L.; Russello, M.A. Population genomics of Sitka black-tailed deer supports invasive species management and ecological restoration on islands. Commun. Biol. 2022, 5, 223. [Google Scholar] [CrossRef]
- Taberlet, P.; Luikart, G. Non-invasive genetic sampling and individual identification. Biol. J. Linn. Soc. 1999, 68, 41–55. [Google Scholar] [CrossRef]
- Moore, S.S.; Sargeant, L.L.; King, T.J.; Mattick, J.S.; Georges, M.; Hetzel, D.J.S. The conservation of dinucleotide microsatellites among mammalian genomes allows the use of heterologous PCR primer pairs in closely related species. Genomics 1991, 10, 654–660. [Google Scholar] [CrossRef]
- Jarne, P.; Lagoda, P.J. Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 1996, 11, 424–429. [Google Scholar] [CrossRef]
- Levý, E.; Putnová, L.; Štohl, R.; Svobodová, K.; Matoušková, J.; Robovský, J.; Lamka, J.; Vrtková, I.; Ernst, M. Utility of several microsatellite markers for the genetic characterisation of three ex situ populations of threatened caprine taxa (Capra aegagrus, C. cylindricornis and C. falconeri). Arch. Anim. Breed. 2015, 58, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.S.; Shih, K.M.; Chang, C.T.; Chung, J.D.; Hwang, S.Y. Testing the effect of mountain ranges as a physical barrier to current gene flow and environmentally dependent adaptive divergence in Cunninghamia konishii (Cupressaceae). Front. Genet. 2019, 10, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waits, L.; Taberlet, P.; Swenson, J.E.; Sandegren, F.; Franzen, R. Nuclear DNA microsatellite analysis of genetic diversity and gene flow in the Scandinavian brown bear (Ursus arctos). Mol. Ecol. 2000, 9, 421–431. [Google Scholar] [CrossRef]
- Hoshino, A.A.; Bravo, J.P.; Nobile, P.M.; Morelli, K.A. Microsatellites as tools for genetic diversity analysis. In Genetic Diversity in Microorganisms; Caliskan, M., Ed.; InTechOpen: London, UK, 2012. [Google Scholar]
- Goleman, M.; Balicki, I.; Radko, A.; Jakubczak, A.; Fornal, A. Genetic diversity of the Polish Hunting Dog population based on pedigree analyses and molecular studies. Livest. Sci. 2019, 229, 114–117. [Google Scholar] [CrossRef]
- Sui, L.; Zhang, F.; Wang, X.; Bossier, P.; Sorgeloos, P.; Hänfling, B. Genetic diversity and population structure of the Chinese mitten crab Eriocheir sinensis in its native range. Mar. Biol. 2009, 156, 1573–1583. [Google Scholar] [CrossRef]
- Bond, J. Microsatellites: Evolution and Applications; Goldstein, D.B., Schlötterer, C., Eds.; Oxford University Press: Oxford, UK, 1999; p. 368. [Google Scholar]
- Queller, D.C.; Strassmann, J.E.; Hughes, C.R. Microsatellites and kinship. Trends Ecol. Evol. 1993, 8, 285–288. [Google Scholar] [CrossRef]
- Pei, J.; Bao, P.; Chu, M.; Liang, C.; Ding, X.; Wang, H.; Wu, X.; Guo, X.; Yan, P. Evaluation of 17 microsatellite markers for parentage testing and individual identification of domestic yak (Bos grunniens). PeerJ 2018, 6, e5946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radko, A.; Podbielska, A. Microsatellite DNA analysis of genetic diversity and parentage testing in the popular dog breeds in Poland. Genes 2021, 12, 485. [Google Scholar] [CrossRef]
- Ciucci, P.; Boitani, L. The Apennine brown bear: A critical review of its status and conservation problems. Ursus 2008, 19, 130–145. [Google Scholar] [CrossRef]
- Peters, W.; Hebblewhite, M.; Cavedon, M.; Pedrotti, L.; Mustoni, A.; Zibordi, F.; Groff, C.; Zanin, M.; Cagnacci, F. Resource selection and connectivity reveal conservation challenges for reintroduced brown bears in the Italian Alps. Biol. Conserv. 2015, 186, 123–133. [Google Scholar] [CrossRef]
- Bombieri, G.; Penteriani, V.; Delgado, M.D.M.; Groff, C.; Pedrotti, L.; Jerina, K. Towards understanding bold behaviour of large carnivores: The case of brown bears in human-modified landscapes. Anim. Conserv. 2021, 24, 783–797. [Google Scholar] [CrossRef]
- Ciucci, P.; Altea, T.; Antonucci, A.; Chiaverini, L.; Di Croce, A.; Mauro, F.; Forconi, P.; Latini, R.; Maiorano, L.; Monaco, A.; et al. Distribution of the brown bear (Ursus arctos marsicanus) in the central Apennines, Italy, 2005–2014. Hystrix 2017, 28, 86. [Google Scholar] [CrossRef]
- Data|portal.mbase.org. Available online: https://portal.mbase.org/db/map/article/29 (accessed on 1 October 2022).
- Scarpulla, E.; Boattini, A.; Cozzo, M.; Giangregorio, P.; Ciucci, P.; Mucci, N.; Randi, E.; Davoli, F. First core microsatellite panel identification in Apennine brown bears (Ursus arctos marsicanus): A collaborative approach. BMC Genom. 2021, 22, 623. [Google Scholar] [CrossRef] [PubMed]
- Davoli, F.; Cozzo, M.; Angeli, F.; Groff, C.; Randi, E. Infanticide in brown bear: A case-study in the Italian Alps-Genetic identification of perpetrator and implications in small populations. Nat. Conserv. 2018, 25, 55–75. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef] [PubMed]
- De Barba, M.; Miquel, C.; Lobréaux, S.; Quenette, P.Y.; Swenson, J.E.; Taberlet, P. High-throughput microsatellite genotyping in ecology: Improved accuracy, efficiency, standardization and success with low-quantity and degraded DNA. Mol. Ecol. Resour. 2017, 17, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Gervasi, V.; Ciucci, P.; Boulanger, J.; Randi, E.; Boitani, L. A multiple data source approach to improve abundance estimates of small populations: The brown bear in the Apennines, Italy. Biol. Conserv. 2012, 152, 10–20. [Google Scholar] [CrossRef]
- Maiorano, L.; Chiaverini, L.; Falco, M.; Ciucci, P. Combining multi-state species distribution models, mortality estimates, and landscape connectivity to model potential species distribution for endangered species in human dominated landscapes. Biol. Conserv. 2019, 237, 19–27. [Google Scholar] [CrossRef]
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Valière, N. GIMLET: A computer program for analysing genetic individual identification data. Mol. Ecol. Notes 2002, 2, 377–379. [Google Scholar] [CrossRef]
- Miller, C.R.; Joyce, P.; Waits, L.P. Assessing allelic dropout and genotype reliability using maximum likelihood. Genetics 2002, 160, 357–366. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smouse, P.E.; Banks, S.C.; Peakall, R. Converting quadratic entropy to diversity: Both animals and alleles are diverse, but some are more diverse than others. PLoS ONE 2017, 12, e0185499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowski, S.T.; Sawaya, M.A.; Taper, M.L. Individual identification and distribution of genotypic differences between individuals. J. Wildl. Manag. 2006, 70, 1148–1150. [Google Scholar] [CrossRef]
- Mills, L.S.; Citta, J.J.; Lair, K.P.; Schwartz, M.K.; Tallmon, D.A. Estimating animal abundance using noninvasive DNA sampling: Promise and pitfalls. Ecol. Appl. 2000, 10, 283–294. [Google Scholar] [CrossRef]
- Waits, L.P.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: Cautions and guidelines. Mol. Ecol. 2001, 10, 249–256. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Woods, J.G.; Paetkau, D.; Lewis, D.; McLellan, B.N.; Proctor, M.; Strobeck, C. Genetic tagging of free-ranging black and brown bears. Wildl. Soc. Bull. 1999, 27, 616–627. [Google Scholar]
- Paetkau, D. An empirical exploration of data quality in DNA-based population inventories. Mol. Ecol. 2003, 12, 1375–1387. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Nagaraju, J.; Reddy, K.D.; Nagaraja, G.M.; Sethuraman, B.N. Comparison of multilocus RFLPs and PCR-based marker systems for genetic analysis of the silkworm, Bombyx mori. Heredity 2001, 86, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Paetkau, D.; Shields, G.F.; Strobeck, C. Gene flow between insular, coastal and interior populations of brown bears in Alaska. Mol. Ecol. 1998, 7, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Paetkau, D.; Calvert, W.; Stirling, I.; Strobeck, C. Microsatellite analysis of population structure in Canadian polar bears. Mol. Ecol. 1995, 4, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Paetkau, D.; Strobeck, C. Microsatellite analysis of genetic variation in black bear populations. Mol. Ecol. 1994, 3, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Bellemain, E.; Taberlet, P. Improved noninvasive genotyping method: Application to brown bear (Ursus arctos) faeces. Mol. Ecol. Notes 2004, 4, 519–522. [Google Scholar] [CrossRef]
- Taberlet, P.; Camarra, J.J.; Griffin, S.; Uhres, E.; Hanotte, O.; Waits, L.P.; Dubois-Paganon, C.; Burke, T.; Bouvet, J. Noninvasive genetic tracking of the endangered Pyrenean brown bear population. Mol. Ecol. 1997, 6, 869–876. [Google Scholar] [CrossRef]
- Breen, M.; Jouquand, S.; Renier, C.; Mellersh, C.S.; Hitte, C.; Holmes, N.G.; Chéron, A.; Suter, N.; Vignaux, F.; Bristow, A.E.; et al. Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Res. 2001, 11, 1784–1795. [Google Scholar] [CrossRef] [Green Version]
- Ostrander, E.A.; Sprague, G.F., Jr.; Rine, J. Identification and characterization of dinucleotide repeat (CA) n markers for genetic mapping in dog. Genomics 1993, 16, 207–213. [Google Scholar] [CrossRef]
- Buono, V.; Galliani, G.; Mancini, E.; Davoli, F.; Mengoni, C.; Mucci, N.; Vignoli, L. An improved microsatellite panel to assess genetic variability of the Italian smooth newt (Lissotriton vulgaris meridionalis). J. Genet. 2018, 97, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Boscari, E.; Marino, I.A.; Caruso, C.; Gessner, J.; Lari, M.; Mugue, N.; Barmintseva, A.; Suciu, R.; Onara, D.; Zane, L.; et al. Defining criteria for the reintroduction of locally extinct populations based on contemporary and ancient genetic diversity: The case of the Adriatic Beluga sturgeon (Huso huso). Divers. Distrib. 2021, 27, 816–827. [Google Scholar] [CrossRef]
- De, R.; Kumar, V.; Ankit, K.; Khan, K.A.; Kumar, H.; Kumar, N.; Habib, B.; Goyal, S.P. Cross-amplification of ungulate microsatellite markers in the endemic Indian antelope or blackbuck (Antilope cervicapra) for population monitoring and conservation genetics studies in south Asia. Mol. Biol. Rep. 2021, 48, 5151–5160. [Google Scholar] [CrossRef]
- McLellan, B.N.; Proctor, M.F.; Huber, D.; Michel, S. Ursus arctos (Amended Version of 2017 Assessment). The IUCN Red List of Threatened Species 2016. Available online: https://www.iucnredlist.org/species/41688/121229971#population (accessed on 21 July 2022).
- Brinkman, T.J.; Schwartz, M.K.; Person, D.K.; Pilgrim, K.L.; Hundertmark, K.J. Effects of time and rainfall on PCR success using DNA extracted from deer fecal pellets. Conserv. Genet. 2010, 11, 1547–1552. [Google Scholar] [CrossRef]
- Marucco, F.; Pletscher, D.H.; Boitani, L.; Schwartz, M.K.; Pilgrim, K.L.; Lebreton, J.D. Wolf survival and population trend using non-invasive capture–recapture techniques in the Western Alps. J. Appl. Ecol. 2009, 46, 1003–1010. [Google Scholar] [CrossRef] [Green Version]
- Gray, T.N.; Vidya, T.N.C.; Potdar, S.; Bharti, D.K.; Sovanna, P. Population size estimation of an Asian elephant population in eastern Cambodia through non-invasive mark-recapture sampling. Conserv. Genet. 2014, 15, 803–810. [Google Scholar] [CrossRef]
- Ghebranious, N.; Vaske, D.; Yu, A.; Zhao, C.; Marth, G.; Weber, J.L. STRP screening sets for the human genome at 5 cM density. BMC Genom. 2003, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Fickel, J.; Hohmann, U. A methodological approach for non-invasive sampling for population size estimates in wild boars (Sus scrofa). Eur. J. Wildl. Res. 2006, 52, 28–33. [Google Scholar] [CrossRef]
- Sikes, R.S.; Gannon, W.L. Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J. Mammal. 2011, 92, 235–253. [Google Scholar] [CrossRef] [Green Version]
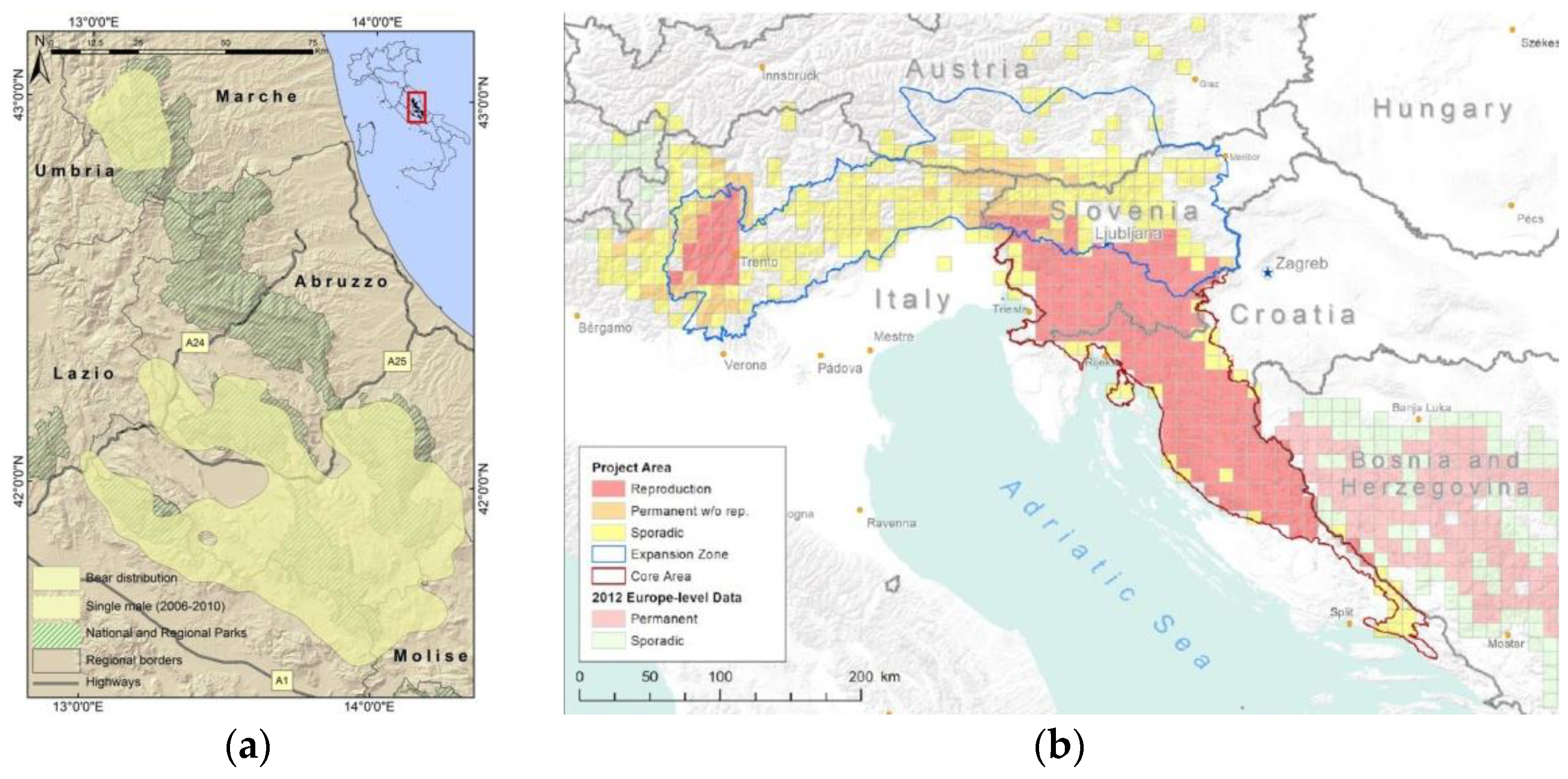
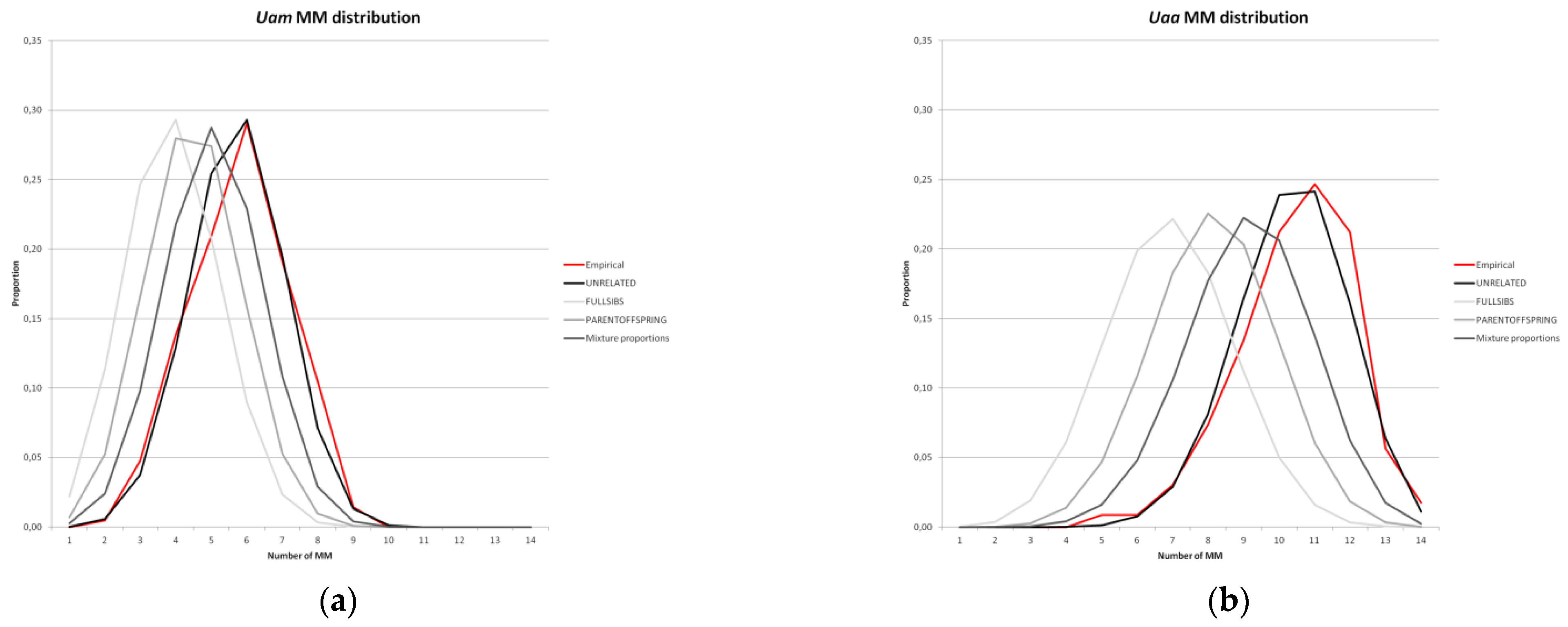


{kind=link}
{kind=link}
{kind=link}
| Subspecies (Abbreviation) | Biological Sample | Sampling Type | Sampling Method | Sex | Genotype ID |
|---|---|---|---|---|---|
| Ursus arctos | Blood | Invasive | Capture and release | F | 73 |
| marsicanus (Uam) | Feces | Non-invasive | Opportunistic | M | 149 |
| Blood | Invasive | Capture and release | F | 31 | |
| Hair | Non-invasive | Opportunistic | M | 81 | |
| Blood | Invasive | Capture and release | F | 99 | |
| Hair | Non-invasive | Opportunistic | M | 127 | |
| Hair | Non-invasive | Opportunistic | M | 128 | |
| Hair | Non-invasive | Opportunistic | M | 135 | |
| Hair | Non-invasive | Opportunistic | M | 142 | |
| Hair | Non-invasive | Rub tree | M | 150 | |
| Hair | Non-invasive | Rub tree | M | 151 | |
| Hair | Non-invasive | Damage | M | 164 | |
| Tissue | Invasive | Carcass | F | 101 | |
| Tissue | Invasive | Carcass | M | 131 | |
| Tissue | Invasive | Carcass | F | 132 | |
| Hair | Non-invasive | Damage | F | 7 | |
| Blood | Invasive | Capture and release | F | 54 | |
| Blood | Invasive | Capture and release | F | 129 | |
| Hair | Non-invasive | Buckthorn patches | M | 10 | |
| Blood | Invasive | Capture and release | M | 66 | |
| Blood | Invasive | Capture and release | M | 83 | |
| Blood | Invasive | Capture and release | M | 117 | |
| Ursus arctos | Hair | Non-invasive | Opportunistic | F | F3 |
| arctos (Uaa) | Hair | Non-invasive | Rub tree | M | M1 |
| Hair | Non-invasive | Opportunistic | F | F23 | |
| Hair | Non-invasive | Opportunistic | F | F2 | |
| Hair | Non-invasive | Opportunistic | M | JJ5 | |
| Hair | Non-invasive | Opportunistic | F | F12 | |
| Hair | Non-invasive | Hair traps | F | F18 | |
| Tissue | Invasive | Opportunistic | F | F4 | |
| Hair | Non-invasive | Rub tree | M | M6 | |
| Hair | Invasive | Capture and release | F | F15 | |
| Bone | Invasive | Carcass | M | M26 | |
| Hair | Non-invasive | Damage | F | Daniza | |
| Hair | Non-invasive | Rub tree | M | Gasper | |
| Hair | Non-invasive | Opportunistic | M | DG2 | |
| Tissue | Invasive | Capture and release | F | DG3 | |
| Hair | Invasive | Capture and release | F | Irma | |
| Hair | Non-invasive | Hair traps | F | Brenta | |
| Hair | Non-invasive | Hair traps | F | Kirka | |
| Feces | Non-invasive | Opportunistic | M | MJ3 | |
| Hair | Non-invasive | Hair traps | M | M4 | |
| Hair | Non-invasive | Hair traps | M | M32 | |
| Hair | Invasive | Opportunistic | M | M33 |
| Locus | Multiplex | Dye | Allele Range | Na | Ne | Ho | UHe | I | HWE | PID | PIDsib | ADO | FA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | |||
| UA03 | 1 | FAM | 100–104 | 96–104 | 2 | 3 | 1.046 | 2.822 | 0.045 | 0.727 | 0.045 | 0.661 | 0.108 | 1.066 | ns | ns | 0.91 | 0.20 | 0.96 | 0.48 | 0.25 | 0 | 0 | 0 |
| UA06 | 1 | HEX | 115–119 | 103–115 | 2 | 3 | 1.046 | 2.051 | 0.045 | 0.455 | 0.045 | 0.524 | 0.108 | 0.860 | ns | ns | 0.91 | 0.31 | 0.96 | 0.57 | 0 | 0 | 0 | 0 |
| UA14 | 2 | HEX | 145–161 | 139–155 | 2 | 4 | 1.963 | 2.969 | 0.591 | 0.727 | 0.502 | 0.679 | 0.684 | 1.207 | ns | ns | 0.38 | 0.17 | 0.60 | 0.46 | 0.17 | 0 | 0 | 0 |
| UA16 | 2 | HEX | 122 | 106–126 | 1 | 6 | 1 | 4.939 | 0 | 0.864 | 0 | 0.816 | 0 | 1.669 | mono | ns | 1 | 0.07 | 1 | 0.37 | 0 | 0 | 0 | 0 |
| UA17 | 1 | FAM | 134 | 138–146 | 1 | 3 | 1 | 2.568 | 0 | 0.545 | 0 | 0.625 | 0 | 1.018 | mono | ns | 1 | 0.22 | 1 | 0.50 | 0 | 0 | 0 | 0 |
| UA25 | 1 | PET | 105–125 | 105–117 | 4 | 3 | 2.696 | 1.678 | 0.773 | 0.318 | 0.644 | 0.413 | 1.136 | 0.726 | ns | ns | 0.20 | 0.39 | 0.48 | 0.65 | 0 | 0.06 | 0 | 0.03 |
| UA51 | 1 | FAM | 120–124 | 112–128 | 2 | 5 | 1.095 | 2.521 | 0.091 | 0.682 | 0.089 | 0.617 | 0.185 | 1.174 | ns | ns | 0.84 | 0.21 | 0.92 | 0.50 | 0 | 0 | 0 | 0 |
| UA57 | 3 | FAM | 108–116 | 112–116 | 2 | 2 | 1.936 | 1.482 | 0.545 | 0.409 | 0.495 | 0.333 | 0.677 | 0.507 | ns | ns | 0.38 | 0.51 | 0.60 | 0.71 | 0.06 | 0 | 0 | 0 |
| UA63 | 2 | NED | 114 | 117–121 | 1 | 3 | 1 | 1.738 | 0 | 0.364 | 0 | 0.434 | 0 | 0.739 | mono | ns | 1 | 0.38 | 1 | 0.63 | 0 | 0 | 0 | 0 |
| UA64 | 2 | PET | 113–121 | 105–109 | 3 | 2 | 2.513 | 1.713 | 0.667 | 0.500 | 0.617 | 0.426 | 1 | 0.607 | ns | ns | 0.23 | 0.43 | 0.51 | 0.65 | 0.04 | 0.02 | 0 | 0 |
| UA65 | 2 | FAM | 127–135 | 123–135 | 2 | 4 | 1.365 | 2.623 | 0.318 | 0.773 | 0.274 | 0.633 | 0.438 | 1.142 | ns | ns | 0.57 | 0.20 | 0.76 | 0.49 | 0.07 | 0.03 | 0 | 0 |
| UA67 | 3 | NED | 124–132 | 124–132 | 3 | 3 | 2.665 | 1.809 | 0.762 | 0.591 | 0.640 | 0.458 | 1.028 | 0.707 | ns | ns | 0.22 | 0.39 | 0.49 | 0.62 | 0.05 | 0.07 | 0 | 0 |
| UA68 | 3 | HEX | 129–141 | 105–137 | 2 | 4 | 1.308 | 2.942 | 0.273 | 0.455 | 0.241 | 0.675 | 0.398 | 1.150 | ns | ns | 0.61 | 0.18 | 0.79 | 0.47 | 0 | 0 | 0 | 0 |
| Locus | Motif Size | Marker Set | Major Allele Frequency | Gene Diversity (GD) | Polymorphic Information Content (PIC) | Allelic Drop-Out (ADO) | False Alleles (FA) | Probability of Identity (PID) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | ||
| G10B 1 | di- | C | - | 0.55 | - | 0.49 | - | 0.37 | - | 0.16 | - | 0 | - | 0.38 | - |
| G10C 1 | di- | C | C | 0.57 | 0.59 | 0.49 | 0.57 | 0.37 | 0.51 | 0 | 0.05 | 0 | 0.01 | 0.38 | 0.24 |
| G10H 1 | di- | - | C | - | 0.84 | - | 0.28 | - | 0.26 | - | 0.07 | - | 0.01 | - | 0.54 |
| G10L 1 | di- | C | C | 0.69 | 0.66 | 0.43 | 0.49 | 0.34 | 0.43 | 0.05 | 0.04 | 0 | 0 | 0.39 | 0.32 |
| G10M 5 | di- | - | C | - | 0.41 | - | 0.68 | - | 0.62 | - | 0.03 | - | 0.01 | - | 0.16 |
| G10P 1 | di- | C | C | 0.81 | 0.30 | 0.31 | 0.75 | 0.26 | 0.70 | 0.08 | 0.05 | 0 | 0.25 | 0.51 | 0.11 |
| G10X 1 | di- | - | C | - | 0.34 | - | 0.75 | - | 0.71 | - | 0.08 | - | 0 | - | 0.10 |
| G1D 1 | di- | C | C | 0.53 | 0.41 | 0.59 | 0.75 | 0.51 | 0.72 | 0.09 | 0.12 | 0 | 0 | 0.30 | 0.10 |
| Mu05 2 | di- | C | - | 0.81 | - | 0.31 | - | 0.26 | - | 0.06 | - | 0 | - | 0.43 | - |
| Mu09 2 | di- | - | C | - | 0.39 | - | 0.75 | - | 0.72 | - | 0.05 | - | 0 | - | 0.10 |
| Mu10 2 | di- | - | C | - | 0.52 | - | 0.61 | - | 0.54 | - | 0.07 | - | 0.12 | - | 0.22 |
| Mu11 2 | di- | C | C | 0.74 | 0.41 | 0.41 | 0.73 | 0.36 | 0.68 | 0.07 | 0.01 | 0 | 0 | 0.33 | 0.12 |
| Mu15 2 | di- | C | C | 0.97 | 0.39 | 0.07 | 0.73 | 0.06 | 0.68 | 0 | 0.04 | 0.04 | 0.03 | 0.77 | 0.12 |
| Mu23 2 | di- | - | C | - | 0.34 | - | 0.72 | - | 0.68 | - | 0.04 | - | 0 | - | 0.12 |
| Mu50 2 | di- | C | C | 0.83 | 0.36 | 0.29 | 0.73 | 0.24 | 0.68 | 0 | 0.03 | 0 | 0 | 0.51 | 0.12 |
| Mu51 2 | di- | C | - | 0.62 | - | 0.48 | - | 0.38 | - | 0.01 | - | 0 | - | 0.37 | - |
| Mu59 2 | di- | C | C | 0.59 | 0.34 | 0.49 | 0.77 | 0.37 | 0.73 | 0 | 0.14 | 0 | 0 | 0.38 | 0.09 |
| cxx20 3 | di- | C | C | 0.46 | 0.34 | 0.64 | 0.73 | 0.57 | 0.68 | 0.09 | 0.32 | 0.02 | 0 | 0.21 | 0.12 |
| REN144A06 3 | di- | C | - | 0.54 | - | 0.57 | - | 0.49 | - | 0.08 | - | 0.04 | - | 0.24 | - |
| UA03 4 | tetra- | N | N | 0.95 | 0.43 | 0.10 | 0.65 | 0.10 | 0.57 | 0.25 | 0 | 0 | 0 | 0.91 | 0.20 |
| UA06 4 | tetra- | N | N | 0.95 | 0.64 | 0.10 | 0.51 | 0.10 | 0.44 | 0 | 0 | 0 | 0 | 0.91 | 0.31 |
| UA14 4 | tetra- | N | N | 0.59 | 0.48 | 0.48 | 0.66 | 0.37 | 0.61 | 0.17 | 0 | 0 | 0 | 0.38 | 0.17 |
| UA16 4 | tetra- | N | N | mono | 0.25 | mono | 0.80 | mono | 0.77 | mono | 0 | mono | 0 | mono | 0.07 |
| UA17 4 | tetra- | N | N | mono | 0.52 | mono | 0.61 | mono | 0.54 | mono | 0 | mono | 0 | mono | 0.22 |
| UA25 4 | tetra- | N | N | 0.54 | 0.75 | 0.63 | 0.40 | 0.57 | 0.37 | 0 | 0.06 | 0 | 0.03 | 0.20 | 0.39 |
| UA51 4 | tetra- | N | N | 0.88 | 0.57 | 0.22 | 0.60 | 0.19 | 0.55 | 0 | 0 | 0 | 0 | 0.84 | 0.21 |
| UA57 4 | tetra- | N | N | 0.59 | 0.80 | 0.48 | 0.33 | 0.37 | 0.27 | 0.06 | 0 | 0 | 0 | 0.38 | 0.51 |
| UA63 4 | tetra- | N | N | mono | 0.73 | mono | 0.42 | mono | 0.38 | mono | 0 | mono | 0 | mono | 0.38 |
| UA64 4 | tetra- | N | N | 0.44 | 0.70 | 0.63 | 0.42 | 0.55 | 0.33 | 0.04 | 0.02 | 0 | 0 | 0.23 | 0.43 |
| UA65 4 | tetra- | N | N | 0.88 | 0.55 | 0.22 | 0.62 | 0.19 | 0.57 | 0.07 | 0.03 | 0 | 0 | 0.57 | 0.20 |
| UA67 4 | tetra- | N | N | 0.48 | 0.68 | 0.62 | 0.45 | 0.54 | 0.37 | 0.05 | 0.07 | 0 | 0 | 0.22 | 0.39 |
| UA68 4 | tetra- | N | N | 0.93 | 0.43 | 0.13 | 0.66 | 0.12 | 0.59 | 0 | 0 | 0 | 0 | 0.61 | 0.18 |
| Mean CURRENT set * | 0.67 | 0.44 | 0.43 | 0.67 | 0.35 | 0.62 | 0.05 | 0.08 | 0.01 | 0.03 | 3.2 × 10−06 | 2.8 × 10−12 | |||
| Mean NEW set * | 0.72 | 0.58 | 0.36 | 0.55 | 0.31 | 0.49 | 0.06 | 0.01 | 0 | 0 | 3.6 × 10−04 | 1.6 × 10−08 | |||
| Mean TOT set * | 0.69 | 0.51 | 0.40 | 0.61 | 0.33 | 0.56 | 0.06 | 0.05 | 0 | 0.02 | 1.1 × 10−09 | 5.2 × 10−21 | |||
| Mean BEST set * | 0.57 | 0.42 | 0.53 | 0.69 | 0.45 | 0.64 | 0.04 | 0.02 | 0 | 0 | 2.0 × 10−05 | 6.9 × 10−11 | |||
| Marker Set * | Ho | UHe | Pairs of Genotypes | PID | PIDsib | EMR | MI | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0MM | 1MM | 2MM | 3MM | |||||||||||||||||
| Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | Uam | Uaa | |
| CURRENT | 0.463 ± 0.050 | 0.694 ± 0.048 | 0.461 ± 0.037 | 0.685 ± 0.035 | 0 | 0 | 0 | 0 | 4.13 × 10−03 | 0 | 4.13 × 10−03 | 0 | 3.2 × 10−06 | 2.8 × 10−12 | 1.9 × 10−03 | 1.5 × 10−05 | 1.90 | 3.34 | 0.67 | 2.08 |
| NEW | 0.316 ± 0.086 | 0.570 ± 0.048 | 0.276 ± 0.074 | 0.561 ± 0.039 | 4.13 × 10−03 | 0 | 6.20 × 10−03 | 0 | 3.51 × 10−02 | 0 | 6.40 × 10−02 | 0 | 3.6 × 10−04 | 1.6 × 10−08 | 2.2 × 10−02 | 3.1 × 10−04 | 2.05 | 2.45 | 0.64 | 1.20 |
| TOT | 0.389 ± 0.051 | 0.636 ± 0.035 | 0.368 ± 0.045 | 0.627 ± 0.028 | 0 | 0 | 0 | 0 | 0 | 0 | 2.07 × 10−03 | 0 | 1.1 × 10−09 | 5.2 × 10−21 | 4.2 × 10−05 | 2.0 × 10−09 | 1.75 | 2.92 | 0.58 | 1.64 |
| BEST | 0.613 ± 0.043 | 0.716 ± 0.038 | 0.545 ± 0.023 | 0.706 ± 0.019 | 0 | 0 | 0 | 0 | 1.65 × 10−02 | 0 | 1.86 × 10−02 | 0 | 2.0 × 10−05 | 6.9 × 10−11 | 5.3 × 10−03 | 5.4 × 10−05 | 2.18 | 3.37 | 0.97 | 2.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buono, V.; Burgio, S.; Macrì, N.; Catania, G.; Hauffe, H.C.; Mucci, N.; Davoli, F. Microsatellite Characterization and Panel Selection for Brown Bear (Ursus arctos) Population Assessment. Genes 2022, 13, 2164. https://doi.org/10.3390/genes13112164
Buono V, Burgio S, Macrì N, Catania G, Hauffe HC, Mucci N, Davoli F. Microsatellite Characterization and Panel Selection for Brown Bear (Ursus arctos) Population Assessment. Genes. 2022; 13(11):2164. https://doi.org/10.3390/genes13112164
Chicago/Turabian StyleBuono, Vincenzo, Salvatore Burgio, Nicole Macrì, Giovanni Catania, Heidi C. Hauffe, Nadia Mucci, and Francesca Davoli. 2022. "Microsatellite Characterization and Panel Selection for Brown Bear (Ursus arctos) Population Assessment" Genes 13, no. 11: 2164. https://doi.org/10.3390/genes13112164