
Complete Mitogenome and Phylogenetic Analyses of Galerita orientalis Schmidt-Goebel, 1846 (Insecta: Coleoptera: Carabidae: Galeritini)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Materials, DNA Extraction, and Illumina Sequencing
2.2. DNA Data Cleaning and Mitogenome Assembly
- (1)
- Trimming adapter sequences with >6 bases;
- (2)
- Removing reads with >0 unidentified nucleotides (Ns);
- (3)
- Removing reads with >20% bases with Phred quality < Q30;
- (4)
- Removing reads with <150 bases.
2.3. Mitogenome Annotation and Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
3.1. Sequencing, Quality Control, and Mitogenome Organization and Base Composition of G. orientalis
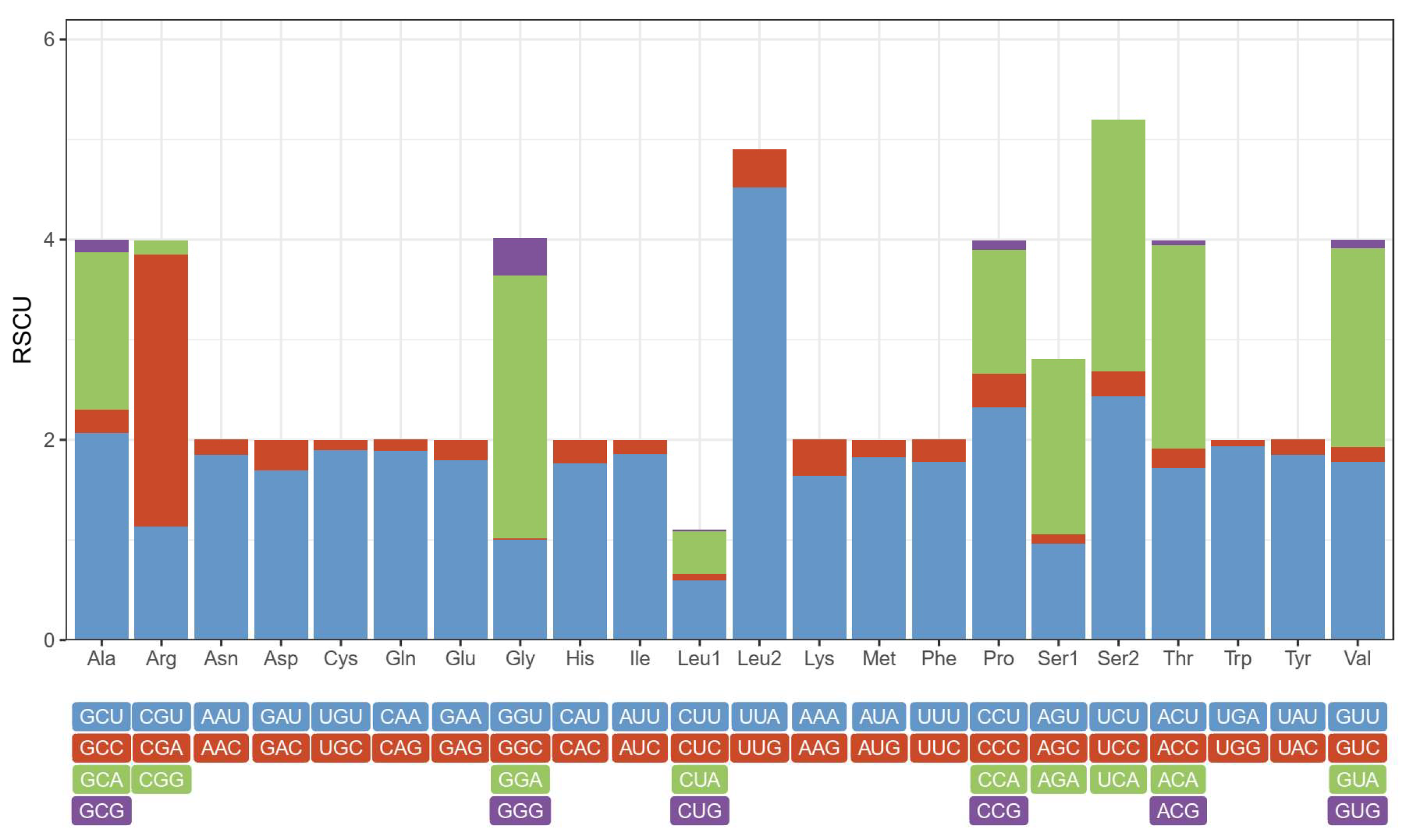
3.2. Protein-Coding Genes
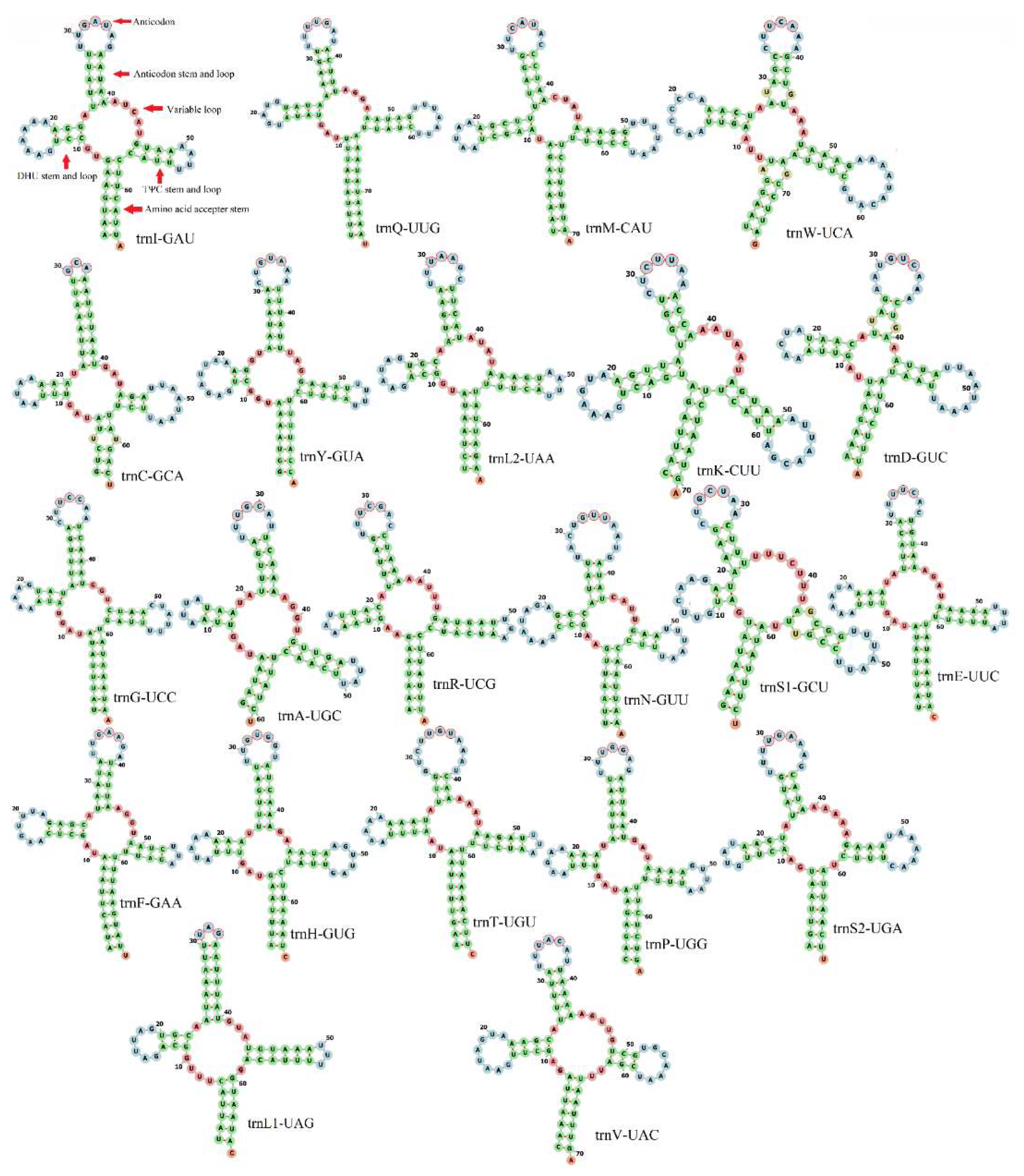
3.3. Transfer and Ribosomal RNA Genes
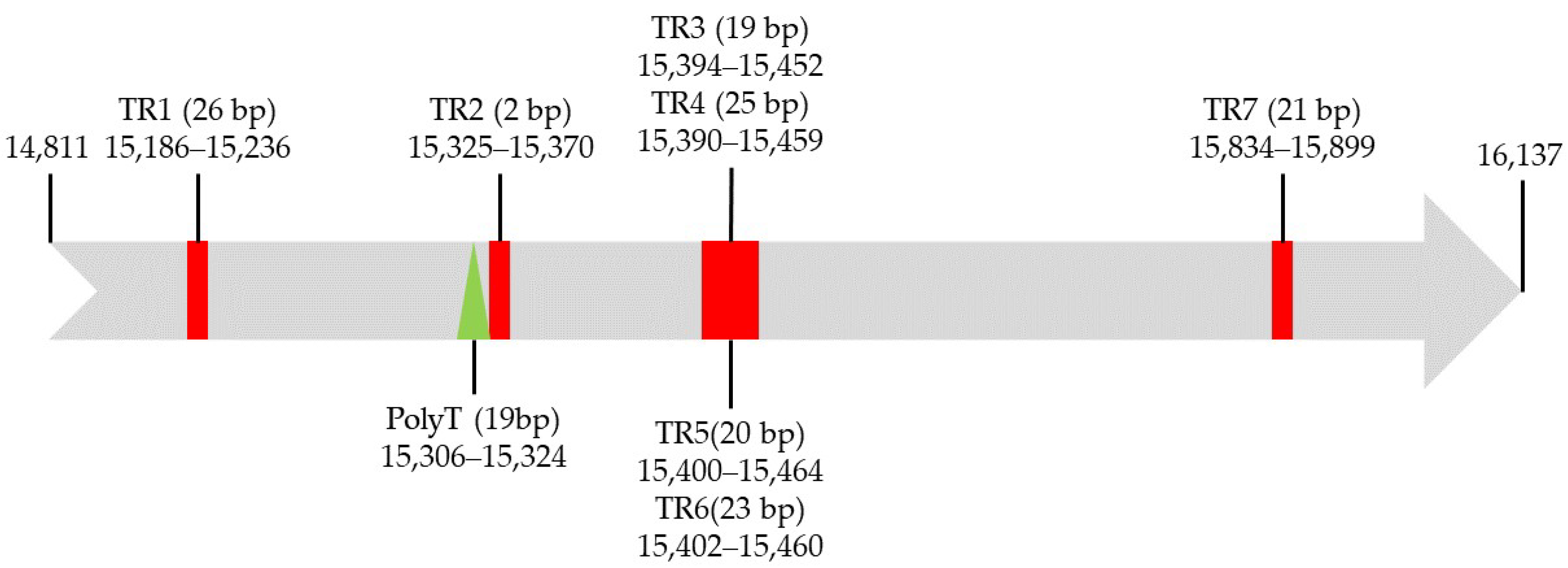
3.4. Control Region
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ober, K.A. Phylogenetic relationships of the carabid subfamily Harpalinae (Coleoptera) based on molecular sequence data. Mol. Phylogenet. Evol. 2002, 24, 228–248. [Google Scholar] [CrossRef]
- Bousquet, Y.; Larochelle, A. Catalogue of the Geadephaga (Coleoptera: Trachypachidae, Rhysodidae, Carabidae including Cicindelini) of America north of Mexico. Mem. Entomol. Soc. Can. 1993, 167, 1–397. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Song, N.; Tang, H.; Yin, X. The Mitochondrial Genome of Amara aulica (Coleoptera, Carabidae, Harpalinae) and Insights into the Phylogeny of Ground Beetles. Genes 2020, 11, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, W. Systematic List of Extant Ground Beetles of the World; W. Lorenz: Tutzing, Germany, 2005. [Google Scholar]
- Ober, K.A.; Maddison, D.R. Phylogenetic relationships of tribes within Harpalinae (Coleoptera: Carabidae) as inferred from 28S ribosomal DNA and the wingless gene. J. Insect Sci. 2008, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovorka, O. A new Galerita species from Bolivia (Coleoptera: Carabidae: Galeritini). Stud. Rep. Taxon. Ser. 2012, 8, 131–134. [Google Scholar]
- Hovorka, O. New species of Galerita Fabricius, 1801 from Panama (Coleoptera: Carabidae: Galeritini). Stud. Rep. Taxon. Ser. 2016, 12, 89–92. [Google Scholar]
- Hovorka, O. Three new Galerita Fabricius, 1801 species (Coleoptera: Carabidae: Galeritini). Stud. Rep. Taxon. Ser. 2017, 13, 323–334. [Google Scholar]
- Hovorka, O. Five new species of Galerita from Asia and new distributional records (Coleoptera: Carabidae: Galeritini). Folia Heyrovskyana Ser. A 2019, 27, 26–41. [Google Scholar]
- Makarov, K.V.; Matalin, A.V. The preimaginal stages of Galerita ruficollis Dejean, 1825 and the position of the tribe Galeritini in the classification of ground beetles (Coleoptera, Carabidae). Zookeys 2021, 1044, 527–561. [Google Scholar] [CrossRef] [PubMed]
- Hunting, W. Female reproductive system of the tribe Galeritini (Coleoptera: Carabidae): Structural features and evolution. Ann. Carnegie Mus. 2008, 77, 229–242, 214. [Google Scholar] [CrossRef]
- Ober, K.A.; Heider, T.N. Phylogenetic diversification patterns and divergence times in ground beetles (Coleoptera: Carabidae: Harpalinae). BMC Evol. Biol. 2010, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, K.A. The Evolution of Arboreal Carabid Beetles; The University of Arizona: Tucson, AZ, USA, 2001. [Google Scholar]
- Liu, Y.-Y.; Zhou, Z.-C.; Chen, X.-S. Characterization of the Complete Mitochondrial Genome of Epicauta impressicornis (Coleoptera: Meloidae) and Its Phylogenetic Implications for the Infraorder Cucujiformia. J. Insect Sci. 2020, 20, 16. [Google Scholar] [CrossRef]
- Linard, B.; Arribas, P.; Andújar, C.; Crampton-Platt, A.; Vogler, A.P. Lessons from genome skimming of arthropod-preserving ethanol. Mol. Ecol. Resour. 2016, 16, 1365–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Tan, W.; Zhang, H.; Jiang, W.; Gao, H.; Wang, W.; Liu, Y.; Wang, Y.; Tian, X. Characterization of the Complete Mitochondrial Genome of Harpalus sinicus and Its Implications for Phylogenetic Analyses. Genes 2019, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Kieran, T.J. Mitochondrial, metagenomic, and phylogenetic analysis of the ground beetle Harpalus pensylvanicus (Coleoptera: Carabidae). Gene 2020, 740, 144540. [Google Scholar] [CrossRef]
- Bai, Y.; Ye, L.; Yang, K.; Wang, H. Genome Survey and SSR Analysis of Camellia nitidissima Chi (Theaceae). Genet. Res. 2022, 2022, 5417970. [Google Scholar] [CrossRef]
- Bai, Y.; Gao, X.; Wang, H.; Ye, L.; Zhang, X.; Huang, W.; Long, X.; Yang, K.; Li, G.; Luo, J.; et al. Comparative mitogenome analysis reveals mitochondrial genome characteristics in eight strains of Beauveria. PeerJ 2022, 10, e14067. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction: Methods and Protocols; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 1–14. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2007, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT—The BLAST-like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2004, 21, 537–539. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [Green Version]
- Gendron, P.; Lemieux, S.; Major, F. Quantitative analysis of nucleic acid three-dimensional structures11Edited by I. Tinoco. J. Mol. Biol. 2001, 308, 919–936. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [Green Version]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the Alignment of Coding Sequences Accounting for Frameshifts and Stop Codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.R.; Kim, M.J.; Jeong, S.Y.; Kim, I. Complete mitochondrial genome sequence of Cicindela anchoralis Chevrolat, 1845 (Coleoptera: Carabidae). Mitochondrial DNA Part B 2018, 3, 282–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-López, A.; Vogler, A.P. The mitogenome phylogeny of Adephaga (Coleoptera). Mol. Phylogenet. Evol. 2017, 114, 166–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.R.; Kim, M.J.; Hong, E.J.; Jeong, J.-C.; Kim, S.S.; Kim, I. Complete mitochondrial genome sequence of Acoptolabrus changeonleei Ishikawa et Kim, 1983 (Coleoptera: Carabidae). Mitochondrial DNA Part B 2019, 4, 1883–1885. [Google Scholar] [CrossRef]
- Liu, N.; Wang, S.; Yang, X.; Song, J.; Wu, J.; Fang, J. The complete mitochondrial genome of Carabus (Damaster) lafossei (Coleoptera: Carabidae). Conserv. Genet. Resour. 2018, 10, 157–160. [Google Scholar] [CrossRef]
- Song, H.; Sheffield, N.C.; Cameron, S.L.; Miller, K.B.; Whiting, M.F. When phylogenetic assumptions are violated: Base compositional heterogeneity and among-site rate variation in beetle mitochondrial phylogenomics. Syst. Entomol. 2010, 35, 429–448. [Google Scholar] [CrossRef]
- Wan, X.; Hong, M.Y.; Liao, A.; Kim, M.I.; Kim, K.-G.; Han, Y.S.; Kim, I. Complete mitochondrial genome of a carabid beetle, Damaster mirabilissimus mirabilissim (Coleoptera: Carabidae). Entomol. Res. 2012, 42, 44–54. [Google Scholar] [CrossRef]
- Timmermans, M.J.T.N.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-Level Sampling of Mitochondrial Genomes in Coleoptera: Compositional Heterogeneity and Phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Raupach, M.J.; Deister, F.; Villastrigo, A.; Balke, M. The complete mitochondrial genomes of Notiophilus quadripunctatus Dejean, 1826 and Omophron limbatum (Fabricius, 1777): New insights into the mitogenome phylogeny of the Carabidae (Insecta, Coleoptera). Insect Syst. Evol. 2022, 53, 242–263. [Google Scholar] [CrossRef]
- Kyndt, E.C.; Kyndt, J.A. Illumina Short-Read Sequencing of the Mitogenomes of Novel Scarites subterraneus Isolates Allows for Taxonomic Refinement of the Genus Scarites Fabricius 1775, within the Carabidae Family. Insects 2022, 13, 190. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, J.; Li, G.; Wang, H.; Luo, J.; Li, C. Complete mitochondrial genome of the common silverfish Lepisma saccharina (Insecta: Zygentoma: Lepismatidae). Mitochondrial DNA Part B 2020, 5, 1552–1553. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Yang, K.; Ye, L.; Gao, X. Complete mitochondrial genome of Pseudoglomeris magnifica (Shelford, 1907) (Insecta: Dictyoptera: Blaberidae). Mitochondrial DNA Part B 2022, 7, 1672–1675. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Species | Whole Length | GenBank Accession Number | Reference |
|---|---|---|---|---|---|
| Carabidae | Brachininae | M. latefasciata | 16,735 bp | ON674050.1 | Unpublished |
| Cicindelinae | Cicindela anchoralis | 16,388 bp | MG253029.1 | [43] | |
| Cicindela puritana | 15,676 bp | MW442537.1 | Unpublished | ||
| Manticora tibialis | 16,439 bp | MF497821.1 | [44] | ||
| Carabinae | Carabus changeonleei | 16,831 bp | MG253028.1 | [45] | |
| Carabus granulatus | 16,918 bp | MN122870.1 | Unpublished | ||
| Carabus lafossei | 16,793 bp | KY992943.1 | [46] | ||
| Calosoma sp. BYU-CO241 | 16,462 bp | GU176340.1 | [47] | ||
| Damaster mirabilissimusmirabilissimus | 16,823 bp | GQ344500.1 | [48] | ||
| Harpalinae | Abax parallelepipedus | 17,701 bp | KT876877.1 | [15] | |
| Amara aulica | 16,646 bp | MN335930.1 | [3] | ||
| Amara communis | 15,745 bp | KX035135.1 | Unpublished | ||
| Craspedophorus nobilis | 15,063 bp | JX412738.1 | [49] | ||
| Galerita orientalis | 16,137 bp | ON920164.1 | This study | ||
| Harpalus anxius | 16,429 bp | ON929899.1 | Unpublished | ||
| Harpalus discrepans | 16,027 bp | OP161482.1 | Unpublished | ||
| Harpalus griseus | 16,972 bp | OP133272.1 | Unpublished | ||
| Harpalus pensylvanicus | 16,434 bp | MN245975.1 | [17] | ||
| Harpalus sinicus | 16,521 bp | MN310888.1 | [16] | ||
| Hexagonia terminalis | 12,639 bp | JX412768.1 | [49] | ||
| Orthomus sp. BMNH 1042407 | 16,368 bp | MK692555.1 | Unpublished | ||
| Pterostichus madidus | 18,324 bp | KT876910.1 | [15] | ||
| Pterostichus niger | 17,160 bp | KT876909.1 | [15] | ||
| Stomis pumicatus | 17,265 bp | KT876914.1 | [15] | ||
| Nebriinae | Nebria brevicollis | 21,161 bp | KT876906.1 | [15] | |
| Notiophilus quadripunctatus | 15,312 bp | MW800883.1 | [50] | ||
| Omophroninae | Omophron limbatum | 15,438 bp | MW800882.1 | [50] | |
| Rhysodinae | Rhysodes sp. BMNH-844233 | 16,315 bp | KX035156.1 | Unpublished | |
| Scaritinae | Scarites subterraneus | 16,163 bp | OL872182.1 | [51] | |
| Trechinae | Tachyta nana | 16,165 bp | KX035142.1 | [15] | |
| Lepismatidae | L. saccharina | 15,244 bp | MT108230.1 | [52] | |
| Blaberidae | Perisphaerinae | Co. magnifica | 16,627 bp | MW630139.1 | [53] |
| Raw Reads Base (bp) | Raw Reads num | Q20 (%) | Q30 (%) | Clean Reads Base (bp) | Clean Reads num | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|---|
| 51,892,104,000 | 345,947,360 | 96.90 | 91.37 | 43,375,436,400 | 289,169,576 | 98.37 | 93.88 | 35.88 |
| Gene | Strand | Location | Size (bp) | Anticodon | Start Codon | Stop Codon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|---|
| trnI | J | 1–65 | 65 | GAT | |||
| trnQ | N | 66–140 | 75 | TTG | 0 | ||
| trnM | J | 136–206 | 71 | CAT | −5 | ||
| nad2 | J | 206–1234 | 1029 | ATA | TAA | −1 | |
| trnW | J | 1232–1306 | 75 | TCA | −3 | ||
| trnC | N | 1333–1396 | 64 | GCA | 2 | ||
| trnY | N | 1398–1465 | 68 | GTA | 1 | ||
| cox1 | J | 1458–2997 | 1540 | ATT | T | −8 | |
| trnL2 | J | 2998–3062 | 65 | TAA | 0 | ||
| cox2 | J | 3065–3749 | 685 | ATG | T | 2 | |
| trnK | J | 3750–3820 | 71 | CTT | 0 | ||
| trnD | J | 3821–3886 | 66 | GTC | 0 | ||
| atp8 | J | 3887–4048 | 162 | ATC | TAA | 0 | |
| atp6 | J | 4042–4719 | 678 | ATG | TAA | −7 | |
| cox3 | J | 4719–5507 | 789 | ATG | TAA | −1 | |
| trnG | J | 5509–5576 | 68 | TCC | 1 | ||
| nad3 | J | 5576–5929 | 354 | ATT | TAG | −1 | |
| trnA | J | 5930–5990 | 61 | TGC | 0 | ||
| trnR | J | 5993–6058 | 66 | TCG | 2 | ||
| trnN | J | 6059–6124 | 66 | GTT | 0 | ||
| trnS1 | J | 6125–6191 | 67 | GCT | 0 | ||
| trnE | J | 6193–6261 | 69 | TTC | 1 | ||
| trnF | N | 6257–6325 | 69 | GAA | −5 | ||
| nad5 | N | 6324–8052 | 1729 | ATT | T | −2 | |
| trnH | N | 8053–8117 | 65 | GTG | 0 | ||
| nad4 | N | 8118–9456 | 1339 | ATG | T | 0 | |
| nad4l | N | 9450–9743 | 294 | ATT | TAA | −7 | |
| trnT | J | 9744–9812 | 69 | TGT | 0 | ||
| trnP | N | 9811–9875 | 65 | TGG | −2 | ||
| nad6 | J | 9877–10,401 | 525 | ATT | TAA | 1 | |
| cob | J | 10,401–11,537 | 1137 | ATG | TAG | −1 | |
| trnS2 | J | 11,536–11,603 | 68 | TGA | −2 | ||
| nad1 | N | 11,620–12,570 | 951 | TTG | TAG | 16 | |
| trnL1 | N | 12,571–12,636 | 66 | TAG | 0 | ||
| rrnL | N | 12,637–13,948 | 1312 | 0 | |||
| trnV | N | 13,954–14,024 | 71 | TAC | 5 | ||
| rrnS | N | 14,025–14,810 | 786 | 0 | |||
| CR | J | 14,811–16,137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Yang, K.; Ye, L.; Gao, X. Complete Mitogenome and Phylogenetic Analyses of Galerita orientalis Schmidt-Goebel, 1846 (Insecta: Coleoptera: Carabidae: Galeritini). Genes 2022, 13, 2199. https://doi.org/10.3390/genes13122199
Bai Y, Yang K, Ye L, Gao X. Complete Mitogenome and Phylogenetic Analyses of Galerita orientalis Schmidt-Goebel, 1846 (Insecta: Coleoptera: Carabidae: Galeritini). Genes. 2022; 13(12):2199. https://doi.org/10.3390/genes13122199
Chicago/Turabian StyleBai, Yu, Kang Yang, Lin Ye, and Xuyuan Gao. 2022. "Complete Mitogenome and Phylogenetic Analyses of Galerita orientalis Schmidt-Goebel, 1846 (Insecta: Coleoptera: Carabidae: Galeritini)" Genes 13, no. 12: 2199. https://doi.org/10.3390/genes13122199