
In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations


Abstract
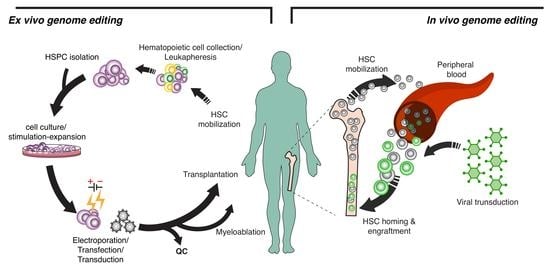
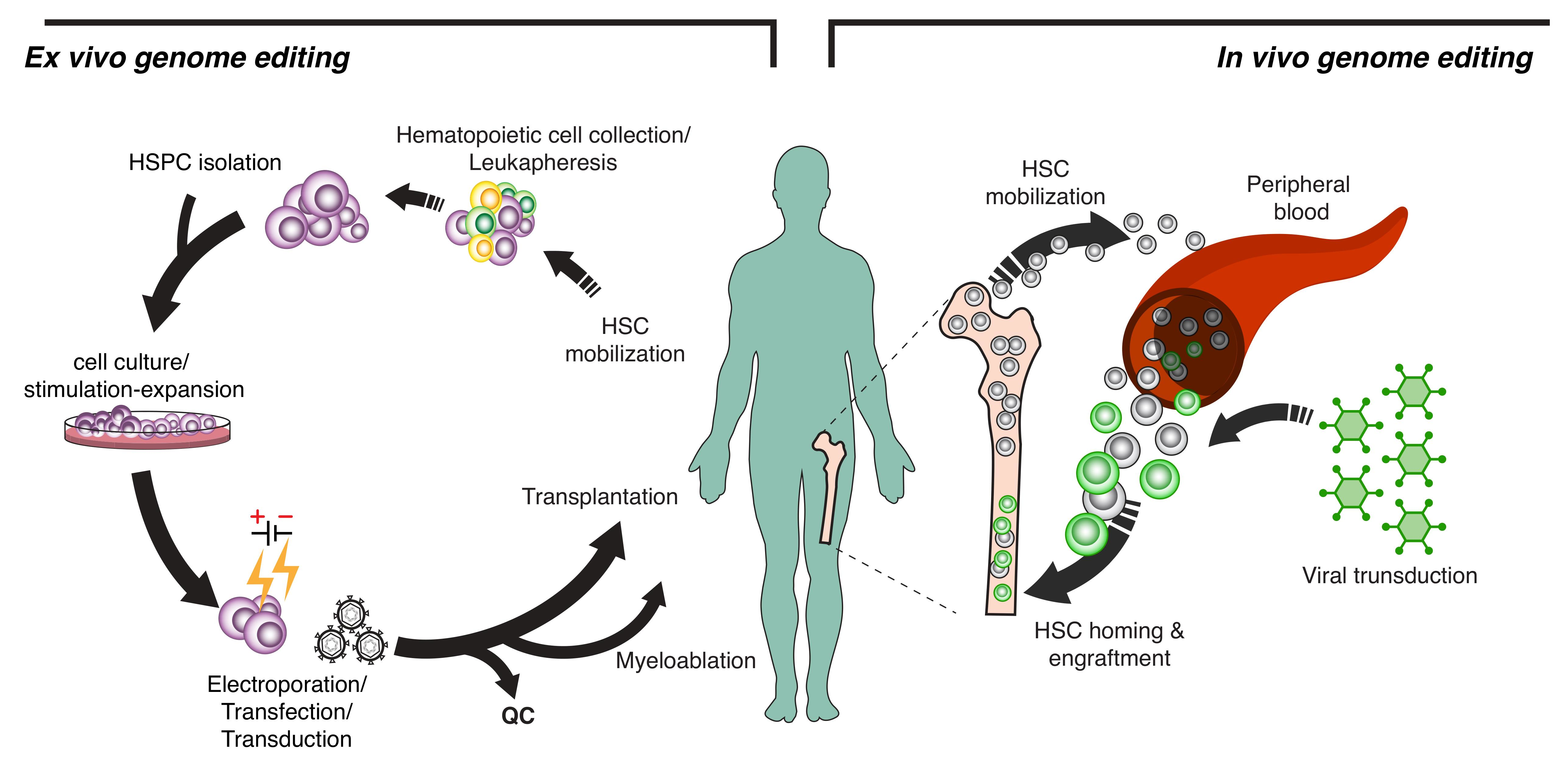
:
{kind=link}
1. Introduction
2. Genome Editing Modules and Ex Vivo HSC Gene Therapy
3. ZFNs
4. TALENS
5. CRISPR-Cas9
6. Double-Strand Break-Free Gene Editing
6.1. Base Editors
6.2. Prime Editing
6.3. Epigenome Editing
6.4. RNA Editing
7. Delivery Tools for In Vivo Gene Therapy
7.1. Lentiviral Vectors
7.2. Adeno-Associated Viral Vectors
7.3. Adenoviral Vectors (Ads)
7.4. Non-Viral Transfer
8. Current Issues and Considerations for In Vivo Gene Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAVs | Adeno-associated viruses |
| ABEs | Adenosine base editors |
| ADAR2 | Adenosine deaminase domain of adenosine deaminase acting on the RNA-2 |
| Ads | Adenoviral vectors |
| CRISPR-Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats CRISPR-Cas9 nuclease |
| CBEs | Cytosine base editors |
| CTL | Cytotoxic T lymphocyte |
| dCas9 | Dead Cas9 |
| DDR | DNA damage response |
| DSBs | Double-strand breaks |
| FA | Fanconi anemia |
| HbF | Fetal hemoglobin |
| HDAd | Helper-dependent adenovirus |
| HSC | Hematopoietic stem cell |
| HSPCs | Hematopoietic stem and progenitor cells |
| HPFH | Hereditary persistence of fetal hemoglobin |
| HDR | Homologous directed repair |
| HIV | Human immunodeficiency virus |
| IAV | Influenza A virus |
| ITRs | Inverted terminal repeats |
| LbL | Layer-by-layer |
| LVs | Lentiviral vectors lvs |
| LCMV | Lymphocytic choriomeningitis virus |
| MHC | Major histocompatibility complex |
| MMEJ | Microhomology-mediated end joining |
| Nab | Neutralizing antibodies |
| NHEJ | Non-homologous end joining |
| PNAs | Peptide nucleic acids |
| PLGA | Poly-lactic-co-glycolic acid |
| pegRNA | Prime editing guide RNA |
| PE | Prime editors |
| PAM | Protospacer adjacent motif |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| ssODN | Single-stranded oligodeoxynucleotide |
| TALENs | Transcription activator-like effector nucleases |
| VOCs | Vaso-occlusive crises |
| VSV | Vesicular stomatitis virus |
| VSV-G | Vesicular stomatitis virus glycoprotein |
| WAS | Wiskott-Aldrich syndrome |
| XLMTM | X-linked myotubular myopathy and Duchenne’s muscular dystrophy |
| X-SCID | X-linked Severe Combined Immunodeficiency |
| ZFNs | Zinc-finger nucleases |
References
- Branzei, D.; Foiani, M. Regulation of DNA Repair throughout the Cell Cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Saleh-Gohari, N.; Helleday, T. Conservative Homologous Recombination Preferentially Repairs DNA Double-Strand Breaks in the S Phase of the Cell Cycle in Human Cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo Genome Editing as a Therapeutic Approach. Int. J. Mol. Sci. 2018, 19, 2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, I.; Flotte, T.R.; Keeler, A.M. CRISPR/Cas-Dependent and Nuclease-Free In Vivo Therapeutic Gene Editing. Hum. Gene Ther. 2021, 32, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-Finger Proteins in Health and Disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; van Attikum, H. DNA Double-Strand Break Repair: Putting Zinc Fingers on the Sore Spot. Semin. Cell Dev. Biol. 2021, 113, 65–74. [Google Scholar] [CrossRef]
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the Sickle Cell Disease Mutation in Human Hematopoietic Stem/Progenitor Cells. Blood 2015, 125, 2597–2604. [Google Scholar] [CrossRef]
- Smith, E.C.; Luc, S.; Croney, D.M.; Woodworth, M.B.; Greig, L.C.; Fujiwara, Y.; Nguyen, M.; Sher, F.; Macklis, J.D.; Bauer, D.E.; et al. Strict in Vivo Specificity of the Bcl11a Erythroid Enhancer. Blood 2016, 128, 2338–2342. [Google Scholar] [CrossRef]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther.-Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.H.; Smith, S.E.; Sullivan, T.; Chen, K.; Zhou, Q.; West, J.A.; Liu, M.; Liu, Y.; Vieira, B.F.; Sun, C.; et al. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther.-Methods Clin. Dev. 2017, 4, 137–148. [Google Scholar] [CrossRef]
- Vierstra, J.; Reik, A.; Chang, K.H.; Stehling-Sun, S.; Zhou, Y.; Hinkley, S.J.; Paschon, D.E.; Zhang, L.; Psatha, N.; Bendana, Y.R.; et al. Functional Footprinting of Regulatory DNA. Nat. Methods 2015, 12, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Sangamo, Sanofi Show Positive Early Data for SCD Gene-Edited Cell Therapy. Available online: https://www.genengnews.com/news/sangamo-sanofi-show-positive-early-data-for-scd-gene-edited-cell-therapy/ (accessed on 21 March 2021).
- Fox, T.A.; Booth, C. Gene Therapy for Primary Immunodeficiencies. Br. J. Haematol. 2021, 193, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Genovese, P.; Schiroli, G.; Escobar, G.; Di Tomaso, T.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted Genome Editing in Human Repopulating Haematopoietic Stem Cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical Modeling Highlights the Therapeutic Potential of Hematopoietic Stem Cell Gene Editing for Correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef]
- Pavel-Dinu, M.; Wiebking, V.; Dejene, B.T.; Srifa, W.; Mantri, S.; Nicolas, C.E.; Lee, C.; Bao, G.; Kildebeck, E.J.; Punjya, N.; et al. Gene Correction for SCID-X1 in Long-Term Hematopoietic Stem Cells. Nat. Commun. 2019, 10, 1634. [Google Scholar] [CrossRef] [Green Version]
- Candotti, F. Gene Therapy for Wiskott-Aldrich Syndrome: Here to Stay. Lancet Haematol. 2019, 6, e230–e231. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, T.J.; Van Caeneghem, Y.; Pourebrahim, R.; Ma, C.; Ni, Z.; Garate, Z.; Crane, A.M.; Li, X.S.; Liao, W.; Gonzalez-Garay, M.; et al. Gene Correction of IPSCs from a Wiskott-Aldrich Syndrome Patient Normalizes the Lymphoid Developmental and Functional Defects. Stem Cell Rep. 2016, 7, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Peterson, C.W.; Wang, J.; Norman, K.K.; Norgaard, Z.K.; Humbert, O.; Tse, C.K.; Yan, J.J.; Trimble, R.G.; Shivak, D.A.; Rebar, E.J.; et al. Long-Term Multilineage Engraftment of Autologous Genome-Edited Hematopoietic Stem Cells in Nonhuman Primates. Blood 2016, 127, 2416–2426. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.K.; Sander, J.D. TALENs: A Widely Applicable Technology for Targeted Genome Editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Boch, J. TALE and TALEN Genome Editing Technologies. Gene Genome Ed. 2021, 2, 100007. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Nain, V. TALENs—an Indispensable Tool in the Era of CRISPR: A Mini Review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lux, C.T.; Pattabhi, S.; Berger, M.; Nourigat, C.; Flowers, D.A.; Negre, O.; Humbert, O.; Yang, J.G.; Lee, C.; Jacoby, K.; et al. TALEN-Mediated Gene Editing of HBG in Human Hematopoietic Stem Cells Leads to Therapeutic Fetal Hemoglobin Induction. Mol. Ther.-Methods Clin. Dev. 2019, 12, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, O.; Peterson, C.W.; Norgaard, Z.K.; Radtke, S.; Kiem, H.P. A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for β-Hemoglobinopathies. Mol. Ther.-Methods Clin. Dev. 2018, 8, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-Thalassemia by CRISPR/Cas- And TALEN-Mediated Disruption of Aberrant Regulatory Elements in Human Hematopoietic Stem and Progenitor Cells. Haematologica 2019, 104, E497–E501. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Tong, Y.; Liu, X.Z.; Wang, T.T.; Cheng, L.; Wang, B.Y.; Lv, X.; Huang, Y.; Liu, D.P. Both TALENs and CRISPR/Cas9 Directly Target the HBB IVS2-654 (C > T) Mutation in β-Thalassemiaderived IPSCs. Sci. Rep. 2015, 5, srep12065. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Cheng, Y.; Lu, D.; Gong, X.; Yang, G.; Gong, Z.; Zhu, Y.; Sang, X.; Fan, S.; Zhang, J.; et al. Treatment of Β654-Thalassaemia by TALENs in a Mouse Model. Cell Prolif. 2018, 51, e12491. [Google Scholar] [CrossRef] [Green Version]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid Regeneration from Gene-Corrected SCID-X1 Subject-Derived IPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Cellectis Presents Initial Preclinical Data on Two Novel Gene Therapies for Patients with RAG1 Severe Combined Immunodeficiency (SCID) and Hyper IgE Syndrome at ESGCT 2021. Available online: https://cellectis.com/en/press/cellectis-presents-initial-preclinical-data-on-two-novel-gene-therapies-for-patients-with-rag1-severe-combined-immunodeficiency-scid-and-hyper-ige-syndrome-at-esgct-2021 (accessed on 12 September 2022).
- Shi, B.; Li, J.; Shi, X.; Jia, W.; Wen, Y. TALEN-Mediated Knockout of CCR5 Confers Protection Against Infection of Human Immunodeficiency Virus. JAIDS J. Acquir. Immune Defic. Syndr. 2017, 74, 229–241. [Google Scholar] [CrossRef]
- Romito, M.; Juillerat, A.; Kok, Y.L.; Hildenbeutel, M.; Rhiel, M.; Andrieux, G.; Geiger, J.; Rudolph, C.; Mussolino, C.; Duclert, A.; et al. Preclinical Evaluation of a Novel TALEN Targeting CCR5 Confirms Efficacy and Safety in Conferring Resistance to HIV-1 Infection. Biotechnol. J. 2021, 16, e2000023. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traxler, E.A.; Yao, Y.; Wang, Y.D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A Genome-Editing Strategy to Treat β-Hemoglobinopathies That Recapitulates a Mutation Associated with a Benign Genetic Condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef]
- Métais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome Editing of HBG1 and HBG2 to Induce Fetal Hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef] [Green Version]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of Fetal Hemoglobin Synthesis by CRISPR/Cas9-Mediated Editing of the Human b-Globin Locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Kamran, S.C.; Orkin, S.H. Reawakening Fetal Hemoglobin: Prospects for New Therapies for the β-Globin Disorders. Blood 2012, 120, 2945–2953. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Xu, S.; Yao, Q.; Zhu, Q.; Kai, Y.; Hsu, J.Y.; Sakon, P.; Pinello, L.; Yuan, G.C.; Bauer, D.E.; et al. Transcription Factor Competition at the γ-Globin Promoters Controls Hemoglobin Switching. Nat. Genet. 2021, 53, 511–520. [Google Scholar] [CrossRef]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Gene Regulation: Transcription Factors LRF and BCL11A Independently Repress Expression of Fetal Hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Lamsfus-Calle, A.; Daniel-Moreno, A.; Antony, J.S.; Epting, T.; Heumos, L.; Baskaran, P.; Admard, J.; Casadei, N.; Latifi, N.; Siegmund, D.M.; et al. Comparative Targeting Analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the Induction of Fetal Hemoglobin. Sci. Rep. 2020, 10, 10133. [Google Scholar] [CrossRef]
- Demirci, S.; Zeng, J.; Wu, Y.; Uchida, N.; Shen, A.H.; Pellin, D.; Gamer, J.; Yapundich, M.; Drysdale, C.; Bonanno, J.; et al. BCL11A Enhancer–Edited Hematopoietic Stem Cells Persist in Rhesus Monkeys without Toxicity. J. Clin. Invest. 2020, 130, 6677–6687. [Google Scholar] [CrossRef] [PubMed]
- Psatha, N.; Georgakopoulou, A.; Li, C.; Nandakumar, V.; Georgolopoulos, G.; Acosta, R.; Paschoudi, K.; Nelson, J.; Chee, D.; Athanasiadou, A.; et al. Enhanced HbF Reactivation by Multiplex Mutagenesis of Thalassemic CD34+ Cells in Vitro and in Vivo. Blood 2021, 138, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly Efficient Therapeutic Gene Editing of Human Hematopoietic Stem Cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Frangoul, H.; Corbacioglu, S.; de la Fuente, J.; Wall, D.; Capellini, M.D.; de Montalembert, M.; Kattamis, A.; Lobitz, S.; Rondelli, D.; et al. Efficacy and Safety of a Single Dose of Ctx001 For Transfusion-Dependent Βeta-Thalassemia And Severe Sickle Cell Disease. In Proceedings of the Conference of European Hematology Association, Vienna, Austria, 9–17 June 2022. [Google Scholar]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 20 June 2022).
- Román-Rodríguez, F.J.; Ugalde, L.; Álvarez, L.; Díez, B.; Ramírez, M.J.; Risueño, C.; Cortón, M.; Bogliolo, M.; Bernal, S.; March, F.; et al. NHEJ-Mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell 2019, 25, 607–621.e7. [Google Scholar] [CrossRef] [Green Version]
- Rai, R.; Romito, M.; Rivers, E.; Turchiano, G.; Blattner, G.; Vetharoy, W.; Ladon, D.; Andrieux, G.; Zhang, F.; Zinicola, M.; et al. Targeted Gene Correction of Human Hematopoietic Stem Cells for the Treatment of Wiskott-Aldrich Syndrome. Nat. Commun. 2020, 11, 4034. [Google Scholar] [CrossRef]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome Editing of CXCR4 by CRISPR/Cas9 Confers Cells Resistant to HIV-1 Infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yao, Y.; Xiao, H.; Li, J.; Liu, Q.; Yang, Y.; Adah, D.; Lu, J.; Zhao, S.; Qin, L.; et al. Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum. Gene Ther. 2018, 29, 51–67. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is Crispr/CAS9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef]
- Schiroli, G.; Conti, A.; Ferrari, S.; della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function Following Transient P53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an On-Target Consequence of CRISPR–Cas9 Genome Editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 Genome Editing Induces Megabase-Scale Chromosomal Truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccaro, M.V.; Xu, J.; Mitchell, C.; Marin, D.; Zimmerman, R.; Rana, B.; Weinstein, E.; King, R.T.; Palmerola, K.L.; Smith, M.E.; et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell 2020, 183, 1650–1664.e15. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Luk, K.; Wolfe, S.A.; Kim, J.S. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu. Rev. Biochem. 2019, 88, 191–220. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, J.; Ge, S.; Lai, L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front. Med. 2021, 8, 649896. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of T to G C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 Nuclease with Expanded Targeting Space. Science 2018, 9, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; Mcclements, M.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic Base Editing of Human Hematopoietic Stem Cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Ma, Y.; Hu, H.; Li, Q.; Yang, Y.; Liu, W.; Yin, S.; Li, W.; Fu, B.; et al. Reactivation of γ-Globin Expression through Cas9 or Base Editor to Treat β-Hemoglobinopathies. Cell Res. 2020, 30, 276–278. [Google Scholar] [CrossRef]
- Antoniou, P.; Hardouin, G.; Martinucci, P.; Frati, G.; Felix, T.; Chalumeau, A.; Fontana, L.; Martin, J.; Masson, C.; Brusson, M.; et al. Base-Editing-Mediated Dissection of a γ -Globin Cis -Regulatory Element for the Therapeutic Reactivation of Fetal Hemoglobin Expression. Nat. Commun. 2022, 13, 6618. [Google Scholar] [CrossRef]
- Newby, G.A.; Yen, J.S.; Woodard, K.J.; Mayuranathan, T.; Lazzarotto, C.R.; Li, Y.; Sheppard-Tillman, H.; Porter, S.N.; Yao, Y.; Mayberry, K.; et al. Base Editing of Haematopoietic Stem Cells Rescues Sickle Cell Disease in Mice. Nature 2021, 595, 295–302. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Mishra, A.; Gil, S.; Hawkins, R.D.; Yannaki, E.; Lieber, A. In Vivo HSPC Gene Therapy with Base Editors Allows for Efficient Reactivation of Fetal G-Globin in b-YAC Mice. Blood Adv. 2021, 5, 1122–1135. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Z.; Sun, H.X.; Ouyang, W.; Dong, G.; Liu, T.; Ge, L.; Zhang, X.; Liu, C.; Gu, Y. Transcriptome Analyses of β-Thalassemia −28(A>G) Mutation Using Isogenic Cell Models Generated by CRISPR/Cas9 and Asymmetric Single-Stranded Oligodeoxynucleotides (AssODNs). Front. Genet. 2020, 11, 577053. [Google Scholar] [CrossRef]
- Gehrke, J.M.; Cervantes, O.; Clement, M.K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An Apobec3a-Cas9 Base Editor with Minimized Bystander and off-Target Activities. Nat. Biotechnol. 2018, 36, 977. [Google Scholar] [CrossRef] [PubMed]
- Knipping, F.; Newby, G.A.; Eide, C.R.; McElroy, A.N.; Nielsen, S.C.; Smith, K.; Fang, Y.; Cornu, T.I.; Costa, C.; Gutierrez-Guerrero, A.; et al. Disruption of HIV-1 Co-Receptors CCR5 and CXCR4 in Primary Human T Cells and Hematopoietic Stem and Progenitor Cells Using Base Editing. Mol. Ther. 2022, 30, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Siegner, S.M.; Clemens, A.; Ugalde, L.; Garcia-Garcia, L.; Bueren, J.A.; Rio, P.; Karasu, M.E.; Corn, J.E. Adenine Base Editing Is an Efficient Approach to Restore Function in FA Patient Cells without Double-Stranded DNA Breaks. bioRxiv 2022, 2022.04.22.489197. [Google Scholar] [CrossRef]
- McAuley, G.E.; Yiu, G.; Newby, G.A.; Kang, S.H.L.; Garibay, A.J.; Butler, J.A.; Christian, V.S.; Fitz-Gibbon, S.; Wong, R.L.; Everette, K.A.; et al. Base Editing of Hematopoietic Stem Cells Rescues T-Cell Development for CD3d Severe Combined Immunodeficiency. In Proceedings of the American Society of Gene and Cell Therapy, Annual Meeting, Washington, DC, USA, 16–19 May 2022. [Google Scholar]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime Editing for Functional Repair in Patient-Derived Disease Models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.-F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced Prime Editing Systems by Manipulating Cellular Determinants of Editing Outcomes. Cell 2021, 184, 5635–5652.e29. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Ferrand, J.; Plessier, A.; Polo, S.E. Control of the Chromatin Response to DNA Damage: Histone Proteins Pull the Strings. Semin. Cell Dev. Biol. 2021, 113, 75–87. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Taylor, E.L.; Westendorf, J.J. Histone Mutations and Bone Cancers. Adv. Exp. Med. Biol. 2021, 1283, 53–62. [Google Scholar] [PubMed]
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S.; Yuan, X.; Rots, M.G.; Strahl, B.D.; Blancafort, P. Epigenetic Reprogramming of Cancer Cells via Targeted DNA Methylation. Epigenetics. 2012, 7, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.L.; Le Lay, J.E.; Ruano, E.G.; Kaestner, K.H. TALE-Mediated Epigenetic Suppression of CDKN2A Increases Replication in Human Fibroblasts. J. Clin. Invest. 2015, 125, 1998–2006. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA Epigenome Editing Using CRISPR-Cas SunTag-Directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.; Hu, R. A CRISPR-Based Approach for Targeted DNA Demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [Green Version]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Núñez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-Epigenetic Modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, Y.; Xiao, Q.; He, B.; Geng, G.; Wang, Z.; Cao, B.; Dong, X.; Bai, W.; Wang, Y.; et al. Programmable RNA Editing with Compact CRISPR–Cas13 Systems from Uncultivated Microbes. Nat. Methods 2021, 18, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA Targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA Editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.; Han, Y.; Wang, Y.; Huang, H.; Qian, P. Programmable System of Cas13-Mediated RNA Modification and Its Biological and Biomedical Applications. Front. Cell Dev. Biol. 2021, 9, 677587. [Google Scholar] [CrossRef]
- Fukuda, M.; Umeno, H.; Nose, K.; Nishitarumizu, A.; Noguchi, R.; Nakagawa, H. Construction of a Guide-RNA for Site-Directed RNA Mutagenesis Utilising Intracellular A-To-I RNA Editing. Sci. Rep. 2017, 7, 41478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freije, C.A.; Myhrvold, C.; Boehm, C.K.; Lin, A.E.; Welch, N.L.; Carter, A.; Metsky, H.C.; Luo, C.Y.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell 2019, 76, 826–837.e11. [Google Scholar] [CrossRef] [Green Version]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876.e12. [Google Scholar] [CrossRef]
- Yin, L.; Zhao, F.; Sun, H.; Wang, Z.; Huang, Y.; Zhu, W.; Xu, F.; Mei, S.; Liu, X.; Zhang, D.; et al. CRISPR-Cas13a Inhibits HIV-1 Infection. Mol. Ther.-Nucleic Acids 2020, 21, 147–155. [Google Scholar] [CrossRef]
- Ide, L.M.; Gangadharan, B.; Chiang, K.Y.; Doering, C.B.; Spencer, H.T. Hematopoietic Stem-Cell Gene Therapy of Hemophilia A Incorporating a Porcine Factor VIII Transgene and Nonmyeloablative Conditioning Regimens. Blood 2007, 110, 2855–2863. [Google Scholar] [CrossRef]
- Naldini, L. Lentiviruses as Gene Transfer Agents for Delivery to Non-Dividing Cells. Curr. Opin. Biotechnol. 1998, 9, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrua, F.; Cicalese, M.P.; Galimberti, S.; Giannelli, S.; Dionisio, F.; Barzaghi, F.; Migliavacca, M.; Bernardo, M.E.; Calbi, V.; Assanelli, A.A.; et al. Lentiviral Haemopoietic Stem/Progenitor Cell Gene Therapy for Treatment of Wiskott-Aldrich Syndrome: Interim Results of a Non-Randomised, Open-Label, Phase 1/2 Clinical Study. Lancet Haematol. 2019, 6, e239–e253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamcarz, E.; Therapy, C.; Zhou, S.; Lockey, T.; Production, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Condori, J.; Dowdy, J.; et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Booth, C.; Shaw, K.L.; Xu-Bayford, J.; Garabedian, E.; Trevisan, V.; Carbonaro-Sarracino, D.A.; Soni, K.; Terrazas, D.; Snell, K.; et al. Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 2021, 384, 2002–2013. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [Green Version]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Bougnres, P.; Schmidt, M.; Von Kalle, C.; Fischer, A.; Cavazzana-Calvo, M.; Aubourg, P. Lentiviral Hematopoietic Cell Gene Therapy for X-Linked Adrenoleukodystrophy. Methods Enzymol. 2012, 507, 187–198. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef]
- Annoni, A.; Goudy, K.; Akbarpour, M.; Naldini, L.; Roncarolo, M.G. Immune Responses in Liver-Directed Lentiviral Gene Therapy. Transl. Res. 2013, 161, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Annoni, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Di Tomaso, T.; Raimondi, A.; Lengler, J.; Holmes, M.C.; Scheiflinger, F.; et al. Genome Editing for Scalable Production of Alloantigen-free Lentiviral Vectors for in Vivo Gene Therapy. EMBO Mol. Med. 2017, 9, 1558–1573. [Google Scholar] [CrossRef] [PubMed]
- DePolo, N.J.; Harkleroad, C.E.; Bodner, M.; Watt, A.T.; Anderson, C.G.; Greengard, J.S.; Murthy, K.K.; Dubensky, T.W.; Jolly, D.J. The Resistance of Retroviral Vectors Produced from Human Cells to Serum Inactivation In Vivo and In Vitro Is Primate Species Dependent. J. Virol. 1999, 73, 6708–6714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.L.; Verhoeyen, E. Baboon Envelope Pseudotyped LVs Outperform VSV-G-LVs for Gene Transfer into Early-Cytokine-Stimulated and Resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.Y.; Schaffer, D.V. Engineering a Serum-Resistant and Thermostable Vesicular Stomatitis Virus G Glycoprotein for Pseudotyping Retroviral and Lentiviral Vectors. Gene Ther. 2013, 20, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Rajawat, Y.S.; Humbert, O.; Cook, S.M.; Radtke, S.; Pande, D.; Enstrom, M.; Wohlfahrt, M.E.; Kiem, H.P. In Vivo Gene Therapy for Canine SCID-X1 Using Cocal-Pseudotyped Lentiviral Vector. Hum. Gene Ther. 2021, 32, 113–127. [Google Scholar] [CrossRef]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Roméro, H.; Selleri, S.; Béland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Guerrero, A.; Cosset, F.L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef]
- Burtner, C.R.; Beard, B.C.; Kennedy, D.R.; Wohlfahrt, M.E.; Adair, J.E.; Trobridge, G.D.; Scharenberg, A.M.; Torgerson, T.R.; Rawlings, D.J.; Felsburg, P.J.; et al. Intravenous Injection of a Foamy Virus Vector to Correct Canine SCID-X1. Blood 2014, 123, 3578–3584. [Google Scholar] [CrossRef] [Green Version]
- Lévy, C.; Amirache, F.; Girard-Gagnepain, A.; Frecha, C.; Roman-Rodríguez, F.J.; Bernadin, O.; Costa, C.; Nègre, D.; Gutierrez-Guerrero, A.; Vranckx, L.S.; et al. Measles Virus Envelope Pseudotyped Lentiviral Vectors Transduce Quiescent Human HSCs at an Efficiency without Precedent. Blood Adv. 2017, 1, 2088–2104. [Google Scholar] [CrossRef]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther.-Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Schokrpur, S.; Archang, M.; Hermann, K.; Sharrow, A.C.; Khanna, P.; Novak, J.; Signoretti, S.; Bhatt, R.S.; Knudsen, B.S.; et al. A Non-Integrating Lentiviral Approach Overcomes Cas9-Induced Immune Rejection to Establish an Immunocompetent Metastatic Renal Cancer Model. Mol. Ther.-Methods Clin. Dev. 2018, 9, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mock, U.; Riecken, K.; Berdien, B.; Qasim, W.; Chan, E.; Cathomen, T.; Fehse, B. Novel Lentiviral Vectors with Mutated Reverse Transcriptase for MRNA Delivery of TALE Nucleases. Sci. Rep. 2014, 4, 6409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett JR, H.A. Alipogene Tiparvovec, an Adeno-Associated Virus Encoding the Ser(447)X Variant of the Human Lipoprotein Lipase Gene for the Treatment of Patients with Lipoprotein Lipase Deficiency. Curr. Opin. Mol. Ther. 2009, 11, 681–691. [Google Scholar] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (AAV2-HRPE65v2) in Patients with RPE65-Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Podsakoff, G.; Wong, K.K.; Chatterjee, S. Efficient Gene Transfer into Nondividing Cells by Adeno-Associated Virus-Based Vectors. J. Virol. 1994, 68, 5656–5666. [Google Scholar] [CrossRef] [Green Version]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Hanawa, H.; Vandergriff, J.; Kelly, P.; Vanin, E.F.; Nienhuis, A.W. Efficient Gene Transfer into Human Cord Blood CD34+ Cells and the CD34+CD38- Subset Using Highly Purified Recombinant Adeno-Associated Viral Vector Preparations That Are Free of Helper Virus and Wild-Type AAV. Gene Ther. 2000, 7, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Santat, L.; Paz, H.; Wong, C.; Li, L.; Macer, J.; Forman, S.; Wong, K.K.; Chatterjee, S. Recombinant AAV2 Transduction of Primitive Human Hematopoietic Stem Cells Capable of Serial Engraftment in Immune-Deficient Mice. Proc. Natl. Acad. Sci. USA 2005, 102, 11053–11058. [Google Scholar] [CrossRef]
- Zhou, S.Z.; Cooper, S.; Kang, L.Y.; Ruggieri, L.; Heimfeld, S.; Srivastava, A.; Broxmeyer, H.E. Adeno-Associated Virus 2-Mediated High Efficiency Gene Transfer into Irm, Nature and Mature Subsets of Hematopoietic Progenitor Cells in Human Umbilical Cord Blood. J. Exp. Med. 1994, 179, 1867–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnazhagan, S.; Mukherjee, P.; Wang, X.S.; Qing, K.; Kube, D.M.; Mah, C.; Kurpad, C.; Yoder, M.C.; Srour, E.F.; Srivastava, A. Adeno-Associated Virus Type 2-Mediated Transduction in Primary Human Bone Marrow-Derived CD34+ Hematopoietic Progenitor Cells: Donor Variation and Correlation of Transgene Expression with Cellular Differentiation. J. Virol. 1997, 71, 8262–8267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrove, P.W.; Vanin, E.F.; Kurtzman, G.J.; Nienhuis, A.W. High-Level Globin Gene Expression Mediated by a Recombinant Adeno- Associated Virus Genome That Contains the 3* g Globin Gene Regulatory Element and Integrates as Tandem Copies in Erythroid Cells. Red Cells 1997, 89, 2167–2175. [Google Scholar] [CrossRef]
- Malik, P.; McQuiston, S.A.; Yu, X.J.; Pepper, K.A.; Krall, W.J.; Podsakoff, G.M.; Kurtzman, G.J.; Kohn, D.B. Recombinant Adeno-Associated Virus Mediates a High Level of Gene Transfer but Less Efficient Integration in the K562 Human Hematopoietic Cell Line. J. Virol. 1997, 71, 1776–1783. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Kauss, M.A.; Kopin, E.; Chandra, M.; Ul-hasan, T.; Miller, E.; Jayandharan, G.R.; Rivers, A.E.; Aslanidi, G.V.; Ling, C.; et al. Optimizing the Transduction Efficiency of Human Hematopoietic Stem Cells Using Capsid-Modified AAV6 Vectors in Vitro and in a Xenograft Mouse Model in Vivo. Cytotherapy 2014, 15, 986–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabhi, S.; Lotti, S.N.; Berger, M.P.; Singh, S.; Lux, C.T.; Jacoby, K.; Lee, C.; Negre, O.; Scharenberg, A.M.; Rawlings, D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther.-Nucleic Acids 2019, 17, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, A.C.; Dever, D.P.; Baik, R.; Camarena, J.; Hsu, I.; Charlesworth, C.T.; Morita, C.; Nakauchi, H.; Porteus, M.H. Cas9-AAV6 Gene Correction of β-Globin in Autologous HSCs Improves Sickle Cell Disease Erythropoiesis in Mice. Nat. Commun. 2021, 12, 686. [Google Scholar] [CrossRef] [PubMed]
- Romero, Z.; Lomova, A.; Said, S.; Miggelbrink, A.; Kuo, C.Y.; Campo-Fernandez, B.; Hoban, M.D.; Masiuk, K.E.; Clark, D.N.; Long, J.; et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019, 27, 1389–1406. [Google Scholar] [CrossRef]
- Wang, J.; Exline, C.M.; Declercq, J.J.; Llewellyn, G.N.; Hayward, B.; Li, P.W.; Shivak, D.A.; Surosky, R.T.; Philip, D.; Holmes, M.C.; et al. Homology-Driven Genome Editing in Hematopoietic Stem and Progenitor Cells Using Zinc Finger Nuclease MRNA and AAV6 Donors. Nat. Biotechnol. 2015, 33, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-Free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828.e5. [Google Scholar] [CrossRef]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J.; Porteus, M.H. A Survey of Ex Vivo/in Vitro Transduction Efficiency of Mammalian Primary Cells and Cell Lines with Nine Natural Adeno-Associated Virus (AAV1-9) and One Engineered Adeno-Associated Virus Serotype. Virol. J. 2013, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Haurigot, V.; Doyon, Y.; Li, T.; Wong, S.Y.; Bhagwat, A.S.; Malani, N.; Anguela, X.M.; Sharma, R.; Ivanciu, L.; et al. In Vivo Genome Editing Restores Hemostasis in a Mouse Model of Hemophilia. Nature 2012, 475, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.X.; Chong, S.; Zhang, H.; Makarova, K.S.; Koonin, E.V.; Cheng, D.R.; Scott, D.A. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 2018, 70, 327–339.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krooss, S.A.; Dai, Z.; Schmidt, F.; Rovai, A.; Fakhiri, J.; Dhingra, A.; Yuan, Q.; Yang, T.; Balakrishnan, A.; Steinbrück, L.; et al. Ex Vivo/In Vivo Gene Editing in Hepatocytes Using “All-in-One” CRISPR-Adeno-Associated Virus Vectors with a Self-Linearizing Repair Template. iScience 2020, 23, 100764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakore, P.I.; Kwon, J.B.; Nelson, C.E.; Rouse, D.C.; Gemberling, M.P.; Oliver, M.L.; Gersbach, C.A. RNA-Guided Transcriptional Silencing in Vivo with S. Aureus CRISPR-Cas9 Repressors. Nat. Commun. 2018, 9, 1674. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In Vivo Genome Editing Using Staphylococcus Aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-Associated Virus Antibody Profiles in Newborns, Children, and Adolescents. Clin. Vaccine Immunol. 2011, 18, 1586–1588. [Google Scholar] [CrossRef] [Green Version]
- Rapti, K.; Louis-Jeune, V.; Kohlbrenner, E.; Ishikawa, K.; Ladage, D.; Zolotukhin, S.; Hajjar, R.J.; Weber, T. Neutralizing Antibodies against AAV Serotypes 1, 2, 6, and 9 in Sera of Commonly Used Animal Models. Mol. Ther. 2012, 20, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Manno, C.S.; Chew, A.J.; Hutchison, S.; Larson, P.J.; Herzog, R.W.; Arruda, V.R.; Tai, S.J.; Ragni, M.V.; Thompson, A.; Ozelo, M.; et al. AAV-Mediated Factor IX Gene Transfer to Skeletal Muscle in Patients with Severe Hemophilia B. Blood 2003, 101, 2963–2972. [Google Scholar] [CrossRef]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming Pre-Existing Humoral Immunity to AAV Using Capsid Decoys. Sci. Transl. Med. 2013, 5, S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, B.H.; A Butler, J.; Felker, G.; Ponikowski, P.; A Voors, A.; Pogoda, J.M.; Provost, R.; Guerrero, J.L.; Hajjar, R.J.; Zsebo, K.M. Prevalence of AAV1 Neutralizing Antibodies and Consequences for a Clinical Trial of Gene Transfer for Advanced Heart Failure. Gene Ther. 2016, 23, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Majowicz, A.; Nijmeijer, B.; Lampen, M.H.; Spronck, L.; de Haan, M.; Petry, H.; van Deventer, S.J.; Meyer, C.; Tangelder, M.; Ferreira, V. Therapeutic HFIX Activity Achieved after Single AAV5-HFIX Treatment in Hemophilia B Patients and NHPs with Pre-Existing Anti-AAV5 NABs. Mol. Ther.-Methods Clin. Dev. 2019, 14, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.M.; Flotte, T.R. Moving Forward after Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P. Astellas Reports Fourth Death in Halted Gene Therapy Trial. Available online: https://www.medscape.com/viewarticle/979152 (accessed on 12 September 2022).
- Burton, K.W. Two Deaths From Liver Failure Linked to Spinal Muscular Atrophy Drug. Available online: https://pharmaphorum.com/news/astellas-reports-fourth-death-in-halted-gene-therapy-trial/ (accessed on 12 September 2022).
- Rowe, W.P.; Huebner, R.J.; Gilmore, L.K.; Parrott, R.H.; Ward, T.G. Isolation of a Cytopathogenic Agent from Human Adenoids LTndergoing Spontaneous Degeneration in Tissue Culture. World Health Organ. Monogr. Ser. 1952, 64, 84. [Google Scholar]
- Rosenfeld, M.A.; Siegfried, W.; Yoshimura, K.; Yoneyama, K.; Fukayama, M.; Stier, L.E.; Pääkkö, P.K.; Gilardi, P.; Stratford-Perricaudet, L.D.; Perricaudet, M.; et al. Adenovirus-Mediated Transfer of a Recombinant A1-Antitrypsin Gene to the Lung Epithelium in Vivo. Science 1991, 252, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.; Hackett, N.R.; El-sawy, T.; Rosengart, T.K.; Hirschowitz, E.A.; Lieberman, M.D.; Lesser, M.L.; Crystal, R.G.; Al, H.E.T. Variability of Human Systemic Humoral Immune Responses to Adenovirus Gene Transfer Vectors Administered to Different Organs. J. Virol. 1999, 73, 6729–6742. [Google Scholar] [CrossRef] [Green Version]
- Crystal, R.G. Adenovirus: The First Effective in Vivo Gene Delivery Vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal Systemic Inflammatory Response Syndrome in a Ornithine Transcarbamylase Deficient Patient Following Adenoviral Gene Transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Fang, B.; Eisensmith, R.C.; Wang, H.; Kay, M.A.; Cross, R.E.; Landen, C.N.; Gordon, G.; Bellinger, D.A.; Read, M.S.; Hu, P.C.; et al. Gene Therpy for Hemophilia B: Host Prolongs the Therapeutic Effect o f Factor I X Immunosuppression. Hum. Gene Ther. 1995, 1044, 1039–1044. [Google Scholar] [CrossRef]
- A Smith, T.; White, B.D.; Gardner, J.M.; Kaleko, M.; McClelland, A. Transient Immunosuppression Permits Successful Repetitive Intravenous Administration of an Adenovirus Vector. Gene Ther. 1996, 3, 496–502. [Google Scholar] [PubMed]
- Goulet, M.A.R.L.Y.N.E.; Gravel, C.; Roy, R.; Tremblay, J.P. Immunosuppression to Control the Immune Reactions Triggered by First-Generation Gene Transfer. Hum. Gene Ther. 1995, 1401, 1391–1401. [Google Scholar]
- Poller, W.; Schneider-Rasp, S.; Liebert, U.; Merklein, F.; Thalheimer, P.; Haack, A.; Schwaab, R.; Schmitt, C.; Brackmann, H.H. Brackmann’ Stabilization of Transgene Expression by Incorporation of E3 Region Genes into an Adenoviral Factor IX Vector and by Transient Anti-CD4 Treatment of the Host. Gene Ther. 1996, 3, 521–530. [Google Scholar] [PubMed]
- Sawchuk, S.J.; Boivin, G.P.; Duwel, L.E.; Ball, W.; Bove, K.; Trapnell, B.; Hirsch, R.; Al, S.E.T. Anti-T Cell Receptor Monoclonal Antibody Transgene Expression Following Gene Prolongs Vivo Adenovirus-Mediated In Synovium Transfer to Mouse Synovium. Hum Gene Ther. 1996, 7, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Cassivi, S.D.; Liu, M.; Boehler, A.; Tanswell, A.K.; Slutsky, A.S.; Keshavjee, S.; Wechsler, A.S.; Rosengart, T.; Carpentier, A.F.; Robbins, R.C. Transgene Expression after Adenovirus-Mediated Retransfection of Rat Lungs Is Increased and Prolonged by Transplant Immunosuppression. J. Thorac. Cardiovasc. Surg. 1999, 117, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Li, Z.; Yumul, R.; Lara, S.; Hemminki, A.; Fender, P.; Lieber, A. Multimerization of Adenovirus Serotype 3 Fiber Knob Domains Is Required for Efficient Binding of Virus to Desmoglein 2 and Subsequent Opening of Epithelial Junctions. J. Virol. 2011, 85, 6390–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 Is a Cellular Receptor for Group B Adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Nilsson, M.; Karlsson, S.; Fan, X. Functionally Distinct Subpopulations of Cord Blood CD34+ Cells Are Transduced by Adenoviral Vectors with Serotype 5 or 35 Tropism. Mol. Ther. 2004, 9, 377–388. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Lieber, A. Efficient Gene Transfer into Human CD34+ Cells by a Retargeted Adenovirus Vector. J. Virol. 2000, 74, 2567–2583. [Google Scholar] [CrossRef] [Green Version]
- Ni, S.; Bernt, K.; Gaggar, A.; Li, Z.Y.; Kiem, H.P.; Lieber, A. Evaluation of Biodistribution and Safety of Adenovirus Vectors Containing Group B Fibers after Intravenous Injection into Baboons. Hum. Gene Ther. 2005, 16, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Alba, R.; Bosch, A.; Chillon, M. Gutless Adenovirus: Last-Generation Adenovirus for Gene Therapy. Gene Ther. 2005, 12, S18–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int. J. Mol. Sci. 2020, 21, 3643. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Villegas, N.; Nurieva, W.; Amberger, M.; Ivics, Z. Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, Piggybac and Tol2 for Genome Engineering. Int. J. Mol. Sci. 2021, 22, 5084. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.P.; Riordan, J.D.; Feddersen, C.R.; Dupuy, A.J. A Hybrid Adenoviral Vector System Achieves Efficient Long-Term Gene Expression in the Liver via PiggyBac Transposition. Hum. Gene Ther. 2015, 26, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Saydaminova, K.; Yumul, R.; Krishnan, R.; Liu, J.; Nagy, E.E.; Singh, M.; Izsvák, Z.; Cattaneo, R.; Uckert, W.; et al. In Vivo Transduction of Primitive Mobilized Hematopoietic Stem Cells after Intravenous Injection of Integrating Adenovirus Vectors. Blood 2016, 128, 2206–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Richter, M.; Psatha, N.; Li, C.; Kim, J.; Liu, J.; Ehrhardt, A.; Nilsson, S.K.; Cao, B.; Palmer, D.; et al. A Combined in Vivo HSC Transduction/Selection Approach Results in Efficient and Stable Gene Expression in Peripheral Blood Cells in Mice. Mol. Ther.-Methods Clin. Dev. 2018, 8, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Psatha, N.; Sgouramali, E.; Gkountis, A.; Siametis, A.; Baliakas, P.; Constantinou, V.; Athanasiou, E.; Arsenakis, M.; Anagnostopoulos, A.; Papayannopoulou, T.; et al. Superior Long-Term Repopulating Capacity of G-CSF + Plerixafor-Mobilized Blood: Implications for Stem Cell Gene Therapy by Studies in the Hbbth-3 Mouse Model. Hum. Gene Ther. Methods 2014, 25, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Yannaki, E.; Karponi, G.; Zervou, F.; Constantinou, V.; Bouinta, A.; Tachynopoulou, V.; Kotta, K.; Jonlin, E.; Papayannopoulou, T.; Anagnostopoulos, A.; et al. Hematopoietic Stem Cell Mobilization for Gene Therapy: Superior Mobilization by the Combination of Granulocyte-Colony Stimulating Factor plus Plerixafor in Patients with β-Thalassemia Major. Hum. Gene Ther. 2013, 24, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Psatha, N.; Yannaki, E.; Athanasiou, E.; Sgouramalli, E.; Mantenoudi, O.; Arsenakis, M.; Anagnostopoulos, A.; Fassas, A. The Combination of AMD3100+G-CSF Successfully Mobilizes HSCs into the Peripheral Blood Compared to G-CSF Alone, in a Thalassemic Mouse Model. Haematol. Hematol. J. 2010, 95, 447. [Google Scholar]
- Karponi, G.; Psatha, N.; Lederer, C.W.; Adair, J.E.; Zervou, F.; Zogas, N.; Kleanthous, M.; Tsatalas, C.; Anagnostopoulos, A.; Sadelain, M.; et al. Plerixafor+G-CSF-Mobilized CD34+ Cells Represent an Optimal Graft Source for Thalassemia Gene Therapy. Blood 2015, 126, 616–619. [Google Scholar] [CrossRef] [Green Version]
- Diana, J.; Manceau, S.; Leblanc, T.; Magnani, A.; Magrin, E.; Bendavid, M.; Couzin, C.; Joseph, L.; Soulier, J.; Cavazzana, M.; et al. A New Step in Understanding Stem Cell Mobilization in Patients with Fanconi Anemia: A Bridge to Gene Therapy. Transfussion 2021, 62, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Tisdale, J.F. Hematopoietic Stem Cell Mobilization with Plerixafor in Sickle Cell Disease. Haematologica 2018, 103, 749–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, N.; Leonard, A.; Stroncek, D.; Panch, S.R.; West, K.; Molloy, E.; Hughes, T.E.; Hauffe, S.; Taylor, T.; Fitzhugh, C.; et al. Safe and Efficient Peripheral Blood Stem Cell Collection in Patients with Sickle Cell Disease Using Plerixafor. Hematologica 2020, 105, e497. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Z.; Li, C.; Gil, S.; Papayannopoulou, T.; Doering, C.B.; Lieber, A. High-Level Protein Production in Erythroid Cells Derived from in Vivo Transduced Hematopoietic Stem Cells. Blood Adv. 2019, 3, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- Humbert, O.; Chan, F.; Rajawat, Y.S.; Torgerson, T.R.; Burtner, C.R.; Hubbard, N.W.; Humphrys, D.; Norgaard, Z.K.; O’Donnell, P.; Adair, J.E.; et al. Rapid Immune Reconstitution of SCID-X1 Canines after G-CSF/AMD3100 Mobilization and in Vivo Gene Therapy. Blood Adv. 2018, 2, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Psatha, N.; Wang, H.; Singh, M.; Samal, H.B.; Zhang, W.; Ehrhardt, A.; Izsvák, Z.; Papayannopoulou, T.; Lieber, A. Integrating HDAd5/35++ Vectors as a New Platform for HSC Gene Therapy of Hemoglobinopathies. Mol. Ther.-Methods Clin. Dev. 2018, 9, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Georgakopoulou, A.; Psatha, N.; Li, C.; Capsali, C.; Samal, H.B.; Anagnostopoulos, A.; Ehrhardt, A.; Izsvák, Z.; Papayannopoulou, T.; et al. In Vivo Hematopoietic Stem Cell Gene Therapy Ameliorates Murine Thalassemia Intermedia. J. Clin. Invest. 2019, 129, 598–615. [Google Scholar] [CrossRef]
- Wang, H.; Georgakopoulou, A.; Li, C.; Liu, Z.; Gil, S.; Bashyam, A.; Yannaki, E.; Anagnostopoulos, A.; Pande, A.; Izsvák, Z.; et al. Curative in Vivo Hematopoietic Stem Cell Gene Therapy of Murine Thalassemia Using Large Regulatory Elements. JCI Insight 2020, 5, e139538. [Google Scholar] [CrossRef]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Wang, H.; Li, C.; Obadan, A.O.; Frizzell, H.; Hsiang, T.-Y.; Gil, S.; Germond, A.; Fountain, C.; Baldessari, A.; Roffler, S.; et al. In Vivo Hematopoietic Stem Cell Gene Therapy for SARS-CoV2 Infection Using a Decoy Receptor. Hum. Gene Ther. 2022, 33, 389–403. [Google Scholar] [CrossRef]
- Saydaminova, K.; Ye, X.; Wang, H.; Richter, M.; Ho, M.; Chen, H.Z.; Xu, N.; Kim, J.S.; Papapetrou, E.; Holmes, M.C.; et al. Efficient Genome Editing in Hematopoietic Stem Cells with Helper-Dependent Ad5/35 Vectors Expressing Site-Specific Endonucleases under MicroRNA Regulation. Mol. Ther.-Methods Clin. Dev. 2015, 2, 14057. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Psatha, N.; Sova, P.; Gil, S.; Wang, H.; Kim, J.; Kulkarni, C.; Valensisi, C.; David Hawkins, R.; Stamatoyannopoulos, G.; et al. Reactivation of G-Globin in Adult b-YAC Mice after Ex Vivo and in Vivo Hematopoietic Stem Cell Genome Editing. Blood 2018, 131, 2915–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Mishra, A.S.; Gil, S.; Wang, M.; Georgakopoulou, A.; Papayannopoulou, T.; Hawkins, R.D.; Lieber, A. Targeted Integration and High-Level Transgene Expression in AAVS1 Transgenic Mice after In Vivo HSC Transduction with HDAd5/35++ Vectors. Mol. Ther. 2019, 27, 2195–2212. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, H.; Gil, S.; Germond, A.; Fountain, C.; Baldessari, A.; Kim, J.; Liu, Z.; Georgakopoulou, A.; Radtke, S.; et al. Safe and Efficient in Vivo Hematopoietic Stem Cell Transduction in Nonhuman Primates Using HDAd5/35++ Vectors. Mol. Ther.-Methods Clin. Dev. 2022, 24, 127–141. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A Multi-Functional AAV-CRISPR-Cas9 and Its Host Response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, C.; Xicluna, R.; Hesnard, L.; Devaux, M.; Jaulin, N.; Guilbaud, M.; Le Duff, J.; Couzinié, C.; Moullier, P.; Saulquin, X.; et al. Tetramer-Based Enrichment of Preexisting Anti-AAV8 CD8+ T Cells in Human Donors Allows the Detection of a TEMRA Subpopulation. Front. Immunol. 2020, 10, 3110. [Google Scholar] [CrossRef]
- Ferla, R.; Alliegro, M.; Dell’Anno, M.; Nusco, E.; Cullen, J.M.; Smith, S.N.; Wolfsberg, T.G.; O’Donnell, P.; Wang, P.; Nguyen, A.D.; et al. Low Incidence of Hepatocellular Carcinoma in Mice and Cats Treated with Systemic Adeno-Associated Viral Vectors. Mol. Ther.-Methods Clin. Dev. 2021, 20, 247–257. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther.-Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Chen, H.; Jin, R.; Weng, T.; Ho, J.K.; You, C.; Zhang, L.; Wang, X.; Han, C. Non-Viral Gene Delivery Systems for Tissue Repair and Regeneration. J. Transl. Med. 2018, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, R.A.; Santo, D.; Farinha, D.; Serra, A.; Faneca, H.; Coelho, J.F.J. High Transfection Efficiency Promoted by Tailor-Made Cationic Tri-Block Copolymer-Based Nanoparticles. Acta Biomater. 2017, 47, 113–123. [Google Scholar] [CrossRef]
- Jung, M.R.; Shim, I.K.; Kim, E.S.; Park, Y.J.; Yang, Y.I.; Lee, S.K.; Lee, S.J. Controlled Release of Cell-Permeable Gene Complex from Poly(L-Lactide) Scaffold for Enhanced Stem Cell Tissue Engineering. J. Control. Release 2011, 152, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Park, T.E.; Singh, B.; Maharjan, S.; Firdous, J.; Cho, M.H.; Kang, S.K.; Yun, C.H.; Choi, Y.J.; Cho, C.S. Major Degradable Polycations as Carriers for DNA and SiRNA. J. Control. Release 2014, 193, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Majidi, A.; Nikkhah, M.; Sadeghian, F.; Hosseinkhani, S. Development of Novel Recombinant Biomimetic Chimeric MPG-Based Peptide as Nanocarriers for Gene Delivery: Imitation of a Real Cargo. Eur. J. Pharm. Biopharm. 2016, 107, 191–204. [Google Scholar] [CrossRef]
- Fernández, E.F.; Santos-Carballal, B.; de Santi, C.; Ramsey, J.M.; MacLoughlin, R.; Cryan, S.A.; Greene, C.M. Biopolymer-Based Nanoparticles for Cystic Fibrosis Lung Gene Therapy Studies. Materials 2018, 11, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, S.C.; Southern, K.W.; Gileadi, U.; Fitzjohn, E.M.; Mofford, K.A.; Waddell, B.E.; Gooi, H.C.; Goddard, C.A.; Hannavy, K.; Smyth, S.E.; et al. Repeat Administration of DNA/Liposomes to the Nasal Epithelium of Patients with Cystic Fibrosis. Gene Ther. 2000, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alton, E.W.F.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated Nebulisation of Non-Viral CFTR Gene Therapy in Patients with Cystic Fibrosis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2b Trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Matsui, H.; Yamamoto, N.; Sato, R.; Munakata, T.; Kohara, M.; Harashima, H. Highly Specific Delivery of SiRNA to Hepatocytes Circumvents Endothelial Cell-Mediated Lipid Nanoparticle-Associated Toxicity Leading to the Safe and Efficacious Decrease in the Hepatitis B Virus. J. Control. Release 2017, 266, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Böttger, R.; Pauli, G.; Chao, P.H.; AL Fayez, N.; Hohenwarter, L.; Li, S.D. Lipid-Based Nanoparticle Technologies for Liver Targeting. Adv. Drug Deliv. Rev. 2020, 154–155, 79–101. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel , J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- McNeer, N.A.; Schleifman, E.B.; Cuthbert, A.; Brehm, M.; Jackson, A.; Cheng, C.; Anandalingam, K.; Kumar, P.; Shultz, L.D.; Greiner, D.L.; et al. Systemic Delivery of Triplex-Forming PNA and Donor DNA by Nanoparticles Mediates Site-Specific Genome Editing of Human Hematopoietic Cells in Vivo. Gene Ther. 2013, 20, 658–669. [Google Scholar] [CrossRef] [Green Version]
- Bahal, R.; Ali McNeer, N.; Quijano, E.; Liu, Y.; Sulkowski, P.; Turchick, A.; Lu, Y.C.; Bhunia, D.C.; Manna, A.; Greiner, D.L.; et al. In Vivo Correction of Anaemia in β-Thalassemic Mice by Γ3PNA-Mediated Gene Editing with Nanoparticle Delivery. Nat. Commun. 2016, 7, 13304. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; van Dijk, T.; Vepris, O.; Li, T.M.W.Y.; Schomann, T.; Baldazzi, F.; Kurita, R.; Nakamura, Y.; Grosveld, F.; Philipsen, S.; et al. PLGA-Nanoparticles for Intracellular Delivery of the CRISPR-Complex to Elevate Fetal Globin Expression in Erythroid Cells. Biomaterials 2021, 268, 120580. [Google Scholar] [CrossRef] [PubMed]
- Cannon, P.; Asokan, A.; Czechowicz, A.; Hammond, P.; Kohn, D.B.; Lieber, A.; Malik, P.; Marks, P.; Porteus, M.; Verhoeyen, E.; et al. Safe and Effective in Vivo Targeting and Gene Editing in Hematopoietic Stem Cells: Strategies for Accelerating Development. Hum. Gene Ther. 2021, 32, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of Preexisting Adaptive Immunity to Cas9 Proteins in Humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-Existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther.-Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.M.; Akyüz, L.; Reinke, P.; Volk, H.D.; Schmueck-Henneresse, M. High Prevalence of Streptococcus Pyogenes Cas9-Reactive T Cells within the Adult Human Population. Nat. Med. 2019, 25, 242–248. [Google Scholar] [CrossRef]
- Ferdosi, S.R.; Ewaisha, R.; Moghadam, F.; Krishna, S.; Park, J.G.; Ebrahimkhani, M.R.; Kiani, S.; Anderson, K.S. Multifunctional CRISPR-Cas9 with Engineered Immunosilenced Human T Cell Epitopes. Nat. Commun. 2019, 10, 1842. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Tanner, M.R.; Lee, C.M.; Hurley, A.E.; De Giorgi, M.; Jarrett, K.E.; Davis, T.H.; Doerfler, A.M.; Bao, G.; Beeton, C.; et al. AAV-CRISPR Gene Editing Is Negated by Pre-Existing Immunity to Cas9. Mol. Ther. 2020, 28, 1432–1441. [Google Scholar] [CrossRef]
- Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 712, 704–712. [Google Scholar]
- Reddy, P.S.; Ganesh, S.; Limbach, M.P.; Brann, T.; Pinkstaff, A.; Kaloss, M.; Kaleko, M.; Connelly, S. Development of Adenovirus Serotype 35 as a Gene Transfer Vector. Virology 2003, 311, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.D.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdary, P.; Shapiro, S.; Makris, M.; Evans, G.; Boyce, S.; Talks, K.; Dolan, G.; Reiss, U.; Phillips, M.; Riddell, A.; et al. Phase 1-2 Trial of AAVS3 Gene Therapy in Patients with Hemophilia B. N. Engl. J. Med. 2022, 387, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Lee, J.; Kim, J.S. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol. Cells 2015, 38, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shengdar, Q.; Tsai, J.K.J. Defining and Improving the Genome-Wide Specificities of CRISPR-Cas9 Nucleases. Nat. Rev. Genet. 2016, 17, 300–312. [Google Scholar] [CrossRef]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In Vivo CRISPR Editing with No Detectable Genome-Wide Offtarget Mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef]
- Atkins, A.; Chung, C.-H.; Allen, A.G.; Dampier, W.; Gurrola, T.E.; Sariyer, I.K.; Nonnemacher, M.R.; Wigdahl, B. Off-Target Analysis in Gene Editing and Applications for Clinical Translation of CRISPR/Cas9 in HIV-1 Therapy. Front. Genome Ed. 2021, 3, 673022. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Nahmad, A.D.; Reuveni, E.; Goldschmidt, E.; Tenne, T.; Liberman, M.; Horovitz-Fried, M.; Khosravi, R.; Kobo, H.; Reinstein, E.; Madi, A.; et al. Frequent Aneuploidy in Primary Human T Cells after CRISPR-Cas9 Cleavage. Nat. Biotechnol. 2022. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Psatha, N.; Paschoudi, K.; Papadopoulou, A.; Yannaki, E. In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations. Genes 2022, 13, 2222. https://doi.org/10.3390/genes13122222
Psatha N, Paschoudi K, Papadopoulou A, Yannaki E. In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations. Genes. 2022; 13(12):2222. https://doi.org/10.3390/genes13122222
Chicago/Turabian StylePsatha, Nikoletta, Kiriaki Paschoudi, Anastasia Papadopoulou, and Evangelia Yannaki. 2022. "In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations" Genes 13, no. 12: 2222. https://doi.org/10.3390/genes13122222
APA StylePsatha, N., Paschoudi, K., Papadopoulou, A., & Yannaki, E. (2022). In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations. Genes, 13(12), 2222. https://doi.org/10.3390/genes13122222