
Bioinformatics Prediction and Machine Learning on Gene Expression Data Identifies Novel Gene Candidates in Gastric Cancer

Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Datasets of Gastric Carcinoma
2.2. Identification of Differentially Expressed Genes
2.3. Gene Set Overrepresentation Analyses
2.4. Reconstruction of Protein–Protein Interaction Network and Identification of Hub Proteins
2.5. Identification of Reporter Transcription Factors and Receptors
2.6. Pre-Clinical Diagnostic Validation of Reporter Biomolecules
2.7. Pre-Clinical Prognostic Validation of Reporter Biomolecules
2.8. Screening the Association of Diagnostic and Prognostic Reporter Biomolecules with Gastric Cancer
2.9. Investigation of Classification Performances of Novel Candidate Biomarkers with Machine Learning Approaches
3. Results
3.1. The Transcriptomic Signatures of Gastric Cancer: Identification of Differentially Expressed Genes
3.2. The Proteomic Signatures of Gastric Cancer: Identification of Hub Proteins
3.3. The Regulatory Signatures of Gastric Cancer: Identification of Reporter Transcription Factors
3.4. The Signaling Signatures of Gastric Cancer: Identification of Reporter Receptors
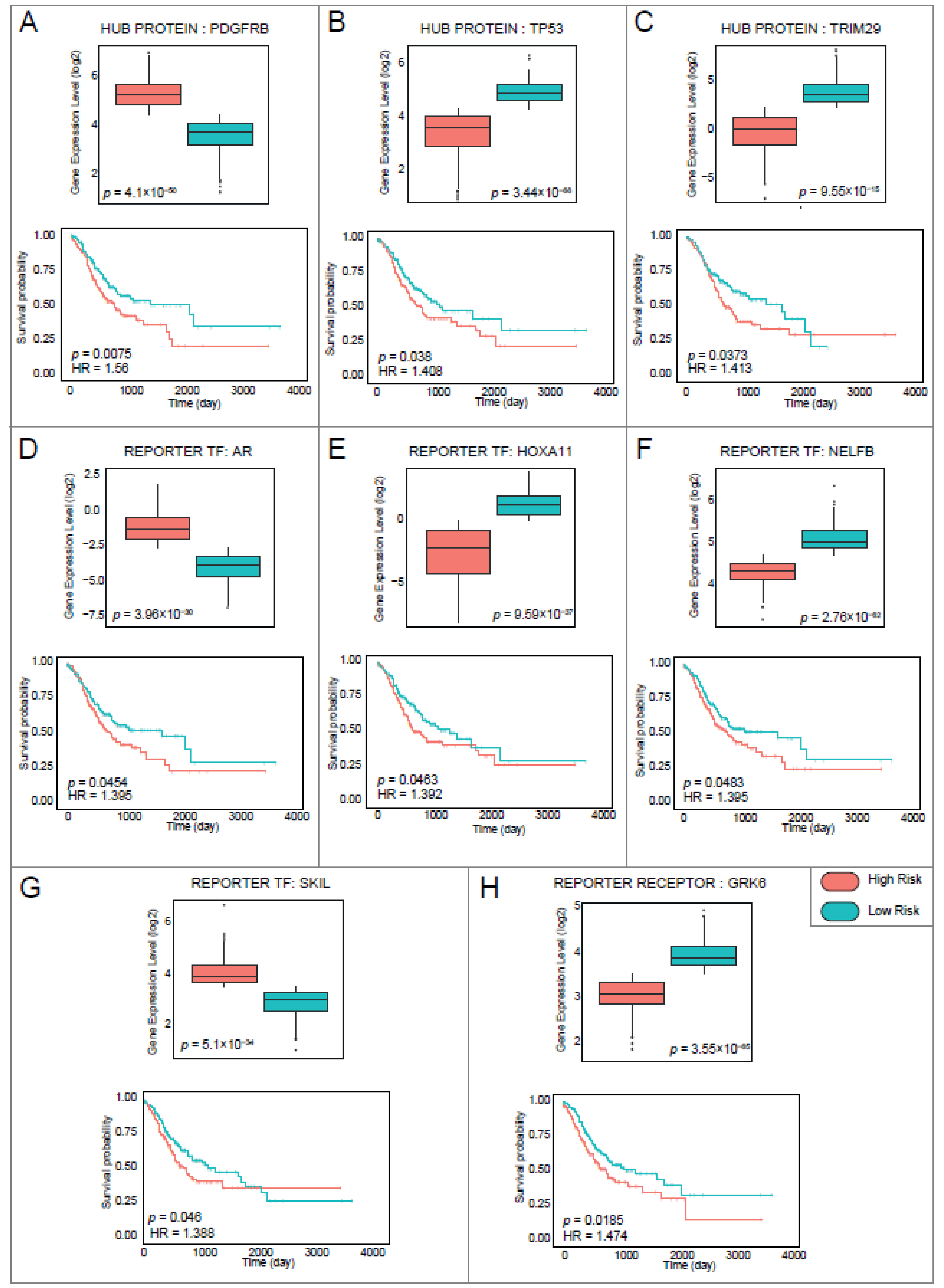
3.5. Diagnostic and Prognostic Power of Reporter Biomolecules of Gastric Cancer
3.6. The Association of Diagnostic and Prognostic Reporter Biomolecules with Gastric Cancer
3.7. Classification Powers of Novel Candidate Biomarkers
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F.J.C. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lyons, K.; Le, L.C.; Pham, Y.T.H.; Borron, C.; Park, J.Y.; Tran, C.T.; Tran, T.V.; Tran, H.T.-T.; Vu, K.T.; Do, C.D.; et al. Gastric cancer: Epidemiology, biology, and prevention: A mini review. Eur. J. Cancer Prev. 2019, 28, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.W.T.; Tan, P. Dissection of gastric cancer heterogeneity for precision oncology. Cancer Sci. 2019, 110, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Biagioni, A.; Skalamera, I.; Peri, S.; Schiavone, N.; Cianchi, F.; Giommoni, E.; Magnelli, L.; Papucci, L. Update on gastric cancer treatments and gene therapies. Cancer Metastasis Rev. 2019, 38, 537–548. [Google Scholar] [CrossRef]
- Correa, R.; Alonso-Pupo, N.; Rodríguez, E.W.H. Multi-omics data integration approaches for precision oncology. Mol. Omics. 2022, 18, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Gulfidan, G.; Soylu, M.; Demirel, D.; Erdonmez, H.B.C.; Beklen, H.; Sarica, P.O.; Arga, K.Y.; Turanli, B. Systems biomarkers for papillary thyroid cancer prognosis and treatment through multi-omics networks. Arch. Biochem. Biophys. 2022, 715, 109085. [Google Scholar] [CrossRef] [PubMed]
- Kelesoglu, N.; Kori, M.; Turanli, B.; Arga, K.Y.; Yilmaz, B.K.; Duru, O.A. Acute Myeloid Leukemia: New Multiomics Molecular Signatures and Implications for Systems Medicine Diagnostics and Therapeutics Innovation. OMICS J. Integr. Biol. 2022, 26, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Kori, M.; Cig, D.; Arga, K.Y.; Kasavi, C. Multiomics Data Integration Identifies New Molecular Signatures for Abdominal Aortic Aneurysm and Aortic Occlusive Disease: Implications for Early Diagnosis, Prognosis, and Therapeutic Targets. OMICS J. Integr. Biol. 2022, 26, 290–304. [Google Scholar] [CrossRef]
- Gov, E. Co-expressed functional module-related genes in ovarian cancer stem cells represent novel prognostic biomarkers in ovarian cancer. Syst. Biol. Reprod. Med. 2020, 66, 255–266. [Google Scholar] [CrossRef]
- Comertpay, B.; Gov, E. Identification of key biomolecules in rheumatoid arthritis through the reconstruction of comprehensive disease-specific biological networks. Autoimmunity 2020, 53, 156–166. [Google Scholar] [CrossRef]
- Rahman, M.R.; Islam, T.; Gov, E.; Turanli, B.; Gulfidan, G.; Shahjaman, M.; Banu, N.A.; Haque, M.; Arga, K.Y.; Moni, M.A. Identification of Prognostic Biomarker Signatures and Candidate Drugs in Colorectal Cancer: Insights from Systems. Biol. Anal. Med. 2019, 55, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kori, M.; Gov, E.; Arga, K.Y. Molecular signatures of ovarian diseases: Insights from network medicine perspective. Syst. Biol. Reprod. Med. 2016, 62, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, J.; Zhang, D.; Bing, Z.; Tian, J.; Ni, M.; Zhang, X.; Meng, Z.; Liu, S. Identification of potential key genes associated with the pathogenesis and prognosis of gastric cancer based on integrated bioinformatics analysis. Front. Genet. 2018, 9, 265. [Google Scholar] [CrossRef]
- Hou, J.Y.; Wang, Y.G.; Ma, S.J.; Yang, B.Y.; Li, Q.P. Identification of a prognostic 5-gene expression signature for gastric cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 619–629. [Google Scholar] [CrossRef]
- Chen, X.; Yang, Y.; Liu, J.; Li, B.; Xu, Y.; Li, C.; Xu, Q.; Liu, G.; Chen, Y.; Ying, J.; et al. Ndrg4 hypermethylation is a potential biomarker for diagnosis and prognosis of gastric cancer in chinese population. Oncotarget 2017, 8, 8105–8119. [Google Scholar] [CrossRef] [Green Version]
- Demirtas, T.Y.; Rahman, M.R.; Yurtsever, M.C.; Gov, E. Forecasting Gastric Cancer Diagnosis, Prognosis, and Drug Repurposing with Novel Gene Expression Signatures. OMICS J. Integr. Biol. 2022, 26, 64–74. [Google Scholar] [CrossRef]
- Wang, Q.; Wen, Y.G.; Li, D.P.; Xia, J.; Zhou, C.-Z.; Wang, D.; Yan, D.-W.; Tang, H.-M.; Peng, Z.H. Upregulated INHBA expression is associated with poor survival in gastric cancer. Med. Oncol. 2012, 29, 77–83. [Google Scholar] [CrossRef]
- Jin, Y.; He, J.; Du, J.; Zhang, R.X.; Yao, H.B.; Shao, Q.S. Overexpression of HS6ST2 is associated with poor prognosis in patients with gastric cancer. Oncol. Lett. 2017, 14, 6191–6197. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 2015, 68–77. [Google Scholar] [CrossRef]
- Kori, M.; Gov, E.; Arga, K.Y. Novel Genomic Biomarker Candidates for Cervical Cancer as Identified by Differential Co-Expression Network Analysis. OMICS J. Integr. Biol. 2019, 23, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Kori, M.; Arga, K.Y.; Mardinoglu, A.; Turanli, B. Repositioning of Anti-Inflammatory Drugs for the Treatment of Cervical Cancer Sub-Types. Front. Pharmacol. 2022, 13, 884548. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of AffymetrixGeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R.; Matthews, L.; Lewis, S.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, D428–D432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Tyers, M. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New Features for Data Integration and Network Visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, K.R.; Nielsen, J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. USA 2005, 102, 2685–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kori, M.; Arga, K.Y. Potential biomarkers and therapeutic targets in cervical cancer: Insights from the meta-analysis of transcriptomics data within network biomedicine perspective. PLoS ONE 2018, 18, e0200717. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Tabas-Madrid, D.; Nogales-Cadenas, R.; Pascual-Montano, A. GeneCodis3: A non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012, 40, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espe, S. Malacards: The Human Disease Database. J. Med. Libr. Assoc. JMLA 2018, 106, 140–141. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García, J.G.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef] [PubMed]
- Schapire, R. Machine Learning Algorithms for Classification; Princeton University: Princeton, NJ, USA, 2015; p. 10. [Google Scholar]
- Kobayashi, H.; Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Hirajima, S.; Miyamae, M.; Okajima, W.; Ohashi, T.; Kosuga, T.; Konishi, H.; et al. Overexpression of denticleless E3 ubiquitin protein ligase homolog (DTL) is related to poor outcome in gastric carcinoma. Oncotarget 2015, 6, 36615–36624. [Google Scholar] [CrossRef] [Green Version]
- Farhadi, J.; Mehrzad, J.; Mehrad-Majd, H.; Motavalizadehkakhky, A. Clinical significance of TRIM29 expression in patients with gastric cancer. Gastroenterol. Hepatol. Bed Bench 2022, 15, 131–138. [Google Scholar]
- Wang, X.; Liu, Y.; Niu, Z.; Fu, R.; Jia, Y.; Zhang, L.; Shao, D.; Du, H.; Hu, Y.; Xing, X.; et al. Prognostic value of a 25-gene assay in patients with gastric cancer after curative resection. Sci. Rep. 2017, 8, 7515. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef]
- Ignatavicius, P.; Dauksa, A.; Zilinskas, J.; Kazokaite, M.; Riauka, R.; Barauskas, G. DNA Methylation of HOXA11 Gene as Prognostic Molecular Marker in Human Gastric Adenocarcinoma. Diagnostics 2022, 12, 1686. [Google Scholar] [CrossRef]
- Gu, H.; Zhong, Y.; Liu, J.; Shen, Q.; Wei, R.; Zhu, H.; Zhang, X.; Xia, X.; Yao, M.; Ni, M. The Role of miR-4256/HOXC8 Signaling Axis in the Gastric Cancer Progression: Evidence From lncRNA-miRNA-mRNA Network Analysis. Front. Oncol. 2022, 11, 793678. [Google Scholar] [CrossRef]
- McChesney, P.A.; Aiyar, S.E.; Lee, O.J.; Zaika, A.; Moskaluk, C.; Li, R.; El-Rifai, W. Cofactor of BRCA1: A novel transcription factor regulator in upper gastrointestinal adenocarcinomas. Cancer Res. 2006, 66, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhang, Z.G. Role of Sp1 expression in gastric cancer: A meta-analysis and bioinformatics analysis. Oncol. Lett. 2019, 18, 4126–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Li, Y.; Cheng, Z.; Wang, P.; Luo, L.; Huang, H.; Duan, S.; Liu, F. GRIK3: A novel oncogenic protein related to tumor TNM stage, lymph node metastasis, and poor prognosis of GC. Tumor Biol. 2017, 39, 1010428317704364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Sharma, P.C. Next generation sequencing-based emerging trends in molecular biology of gastric cancer. Am. J. Cancer Res. 2018, 8, 207–225. [Google Scholar] [PubMed]
- Dong, X.; Wang, G.; Zhang, G.; Ni, Z.; Suo, J.; Cui, J.; Cui, A.; Yang, Q.; Xu, Y.; Li, F. The endothelial lipase protein is promising urinary biomarker for diagnosis of gastric cancer. Diagn. Pathol. 2013, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, A.; Hagström, J.; Mustonen, H.; Kokkola, A.; Tervahartiala, T.; Sorsa, T.; Böckelman, C.; Haglund, C. Serum MMP-8 and TIMP-1 as prognostic biomarkers in gastric cancer. Tumor Biol. 2018, 40, 1010428318799266. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Zheng, K.; Wu, Y.; Wang, O. Pannexin 1 Mediates Gastric Cancer Cell Epithelial-Mesenchymal Transition via Aquaporin 5. Biol. Pharm. Bull. 2021, 44, 1111–1119. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–2040: A population-based modelling study. EClinical Med. 2022, 47, 101404. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Ji, C.D.; Xiao, H.L.; Zhao, H.B.; Cui, Y.H.; Bian, X.W. Reorganized Collagen in the Tumor Microenvironment of Gastric Cancer and Its Association with Prognosis. J. Cancer 2017, 8, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Beagle, B.; Johnson, G.V. AES/GRG5: More than just a dominant-negative TLE/GRG family member. Dev. Dyn. 2010, 239, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Sonoshita, M.; Kakizaki, F.; Aoyama, N.; Itatani, Y.; Uegaki, M.; Sakamoto, H.; Kobayashi, T.; Inoue, T.; Kamba, T.; et al. Amino-terminal enhancer of split gene AES encodes a tumor and metastasis suppressor of prostate cancer. Cancer Sci. 2017, 108, 744–752. [Google Scholar] [CrossRef]
- Kakizaki, F.; Sonoshita, M.; Miyoshi, H.; Itatani, Y.; Ito, S.; Kawada, K.; Taketo, M.M. Expression of metastasis suppressor gene AES driven by a Yin Yang (YY) element in a CpG island promoter and transcription factor YY2. Cancer Sci. 2016, 107, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.M.; Qian, J.; Lin, J.; Wang, Y.-L.; Chen, Q.; Qian, Z.; Li, Y.; Wang, C.-Z.; Yang, J. Aberrant methylation of CCAAT/enhancer binding protein zeta promoter in acute myeloid leukemia. Leuk. Res. 2011, 35, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, L.; Shen, Z.; Li, Y.; Cao, H.; Peng, L.; Qiu, Y.; Cheng, X.; Meng, M.; Lu, D.; et al. CEBPG promotes esophageal squamous cell carcinoma progression by enhancing PI3K-AKT signaling. Am. J. Cancer Res. 2020, 10, 3328–3344. [Google Scholar] [PubMed]
- Stegen, M.; Engler, A.; Ochsenfarth, C.; Manthey, I.; Peters, J.; Siffert, W.; Frey, U.H. Characterization of the G protein-coupled receptor kinase 6 promoter reveals a functional CREB binding site. PLoS ONE 2021, 16, e0247087. [Google Scholar] [CrossRef]
- Tao, R.; Li, Q.; Gao, X.; Ma, L. Overexpression of GRK6 associates with the progression and prognosis of colorectal carcinoma. Oncol. Lett. 2018, 15, 5879–5886. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Zhong, L.; Liu, J.; Feng, J.; Bian, T.; Zhang, Q.; Chen, J.; Lv, X.; Chen, J.; Liu, Y. Prognostic value of decreased GRK6 expression in lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 2541–2549. [Google Scholar] [CrossRef]
- Li, Y.P. GRK6 expression in patients with hepatocellular carcinoma. Asian Pac. J. Trop. Med. 2013, 6, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Zhang, H.; Liu, J.; Rubin, J.B.; Cho, Y.J.; Shu, H.K.; Schniederjan, M.; MacDonald, T.J. Growth factor receptor-Src-mediated suppression of GRK6 dysregulates CXCR4 signaling and promotes medulloblastoma migration. Mol. Cancer 2013, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.J.; Oh, D.K. Prostaglandin synthases: Molecular characterization and involvement in prostaglandin biosynthesis. Prog. Lipid Res. 2017, 66, 50–68. [Google Scholar] [CrossRef]
- Chung, C.C.; Kanetsky, P.A.; Wang, Z.; Hildebrandt, M.A.; Koster, R.; Skotheim, R.I.; Kratz, C.P.; Turnbull, C.; Cortessis, V.K.; Bakken, A.C.; et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 2013, 45, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Luo, T.; Li, Q.; Xu, L.; Xie, Y. HPGDS is a novel prognostic marker associated with lipid metabolism and aggressiveness in lung adenocarcinoma. Front. Oncol. 2022, 12, 5788. [Google Scholar]
- Deheuninck, J.; Luo, K. Ski and SnoN potent negative regulators of TGF-beta signaling. Cell Res. 2009, 19, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Akagi, I.; Miyashita, M.; Makino, H.; Nomura, T.; Hagiwara, N.; Takahashi, K.; Tajiri, T. SnoN overexpression is predictive of poor survival in patients with esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2008, 15, 2965–2975. [Google Scholar] [CrossRef]
- Hagerstrand, D.; Tong, A.; Schumacher, S.E.; Ilic, N.; Shen, R.R.; Cheung, H.W.; Hahn, W.C. Systematic Interrogation of 3q26 Identifies TLOC1 and SKIL as Cancer DriversTLOC1 and SKIL as Cancer Drivers in 3q26. Cancer Discov. 2013, 3, 1044–1057. [Google Scholar] [CrossRef] [Green Version]
- Raffoul, F.; Campla, C.; Nanjundan, M. SnoN/SkiL, a TGFbeta signaling mediator: A participant in autophagy induced by arsenic trioxide. Autophagy 2010, 6, 955–957. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.M.; Patel, S.; Raffoul, F.; Haller, E.; Mills, G.B.; Nanjundan, M. Arsenic trioxide induces a beclin-1-independent autophagic pathway via modulation of SnoN/SkiL expression in ovarian carcinoma cells. Cell Death Differ. 2010, 17, 1867–1881. [Google Scholar] [CrossRef]
- Lazarus, K.A.; Hadi, F.; Zambon, E.; Bach, K.; Santolla, M.F.; Watson, J.K.; Khaled, W.T. BCL11A interacts with SOX2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat. Commun. 2018, 9, 3327. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Davie, J.R. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat. 2010, 192, 275–283. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, L.; Shen, S.; Li, J.; Lu, H.; Mo, W.; Feng, Z. Sp1 cooperates with Sp3 to upregulate MALAT1 expression in human hepatocellular carcinoma. Oncol. Rep. 2015, 34, 2403–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajita, Y.; Kato, T., Jr.; Tamaki, S.; Furu, M.; Takahashi, R.; Nagayama, S.; Toguchida, J. The transcription factor Sp3 regulates the expression of a metastasis-related marker of sarcoma, actin filament-associated protein 1-like 1 (AFAP1L1). PLoS ONE 2013, 8, e49709. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Name | Diagnostic? | Prognostic? | Association with Gastric Cancer |
|---|---|---|---|---|
| Hub Protein | DTL | + | − | [46] |
| FN1 | + | − | [42,43] | |
| PDGFRB | + | + | [42,43] | |
| TP53 | + | + | [42,43,44] | |
| TRIM29 | + | + | [47] | |
| Reporter Transcription Factor | AES | + | − | Novel |
| AR | + | + | [42,43] | |
| CEBPZ | + | − | Novel | |
| GZF1 | + | − | [48] | |
| HDAC2 | + | − | [49] | |
| HOXA11 | + | + | [50] | |
| HOXC8 | + | − | [51] | |
| NELFB | + | + | [52] | |
| NFKB1 | + | − | [42,43] | |
| SKIL | + | + | Novel | |
| SP1 | + | − | [53] | |
| SP3 | + | − | Novel | |
| Reporter Receptor | ATP4A | + | − | [42] |
| BUB1 | + | − | [42] | |
| GRIK3 | + | − | [54] | |
| GRK6 | − | + | Novel | |
| GRM8 | + | − | [55] | |
| HPGDS | + | − | Novel | |
| LIPG | + | − | [56] | |
| MMP1 | + | − | [42,43] | |
| MMP14 | + | − | [42,43] | |
| MMP3 | + | − | [42,43] | |
| MMP8 | + | − | [57] | |
| MMP9 | + | − | [42,43] | |
| NOS3 | + | − | [42,43,44] | |
| PANX1 | + | − | [58] | |
| SRC | + | − | [42,43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kori, M.; Gov, E. Bioinformatics Prediction and Machine Learning on Gene Expression Data Identifies Novel Gene Candidates in Gastric Cancer. Genes 2022, 13, 2233. https://doi.org/10.3390/genes13122233
Kori M, Gov E. Bioinformatics Prediction and Machine Learning on Gene Expression Data Identifies Novel Gene Candidates in Gastric Cancer. Genes. 2022; 13(12):2233. https://doi.org/10.3390/genes13122233
Chicago/Turabian StyleKori, Medi, and Esra Gov. 2022. "Bioinformatics Prediction and Machine Learning on Gene Expression Data Identifies Novel Gene Candidates in Gastric Cancer" Genes 13, no. 12: 2233. https://doi.org/10.3390/genes13122233