
Genome-Wide Identification and Analysis of DOF Gene Family in Eugenia uniflora L. (Myrtaceae)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of DOF Genes in Eugenia uniflora
2.2. In Silico Predictions
2.3. Alignment and Phylogeny Reconstruction
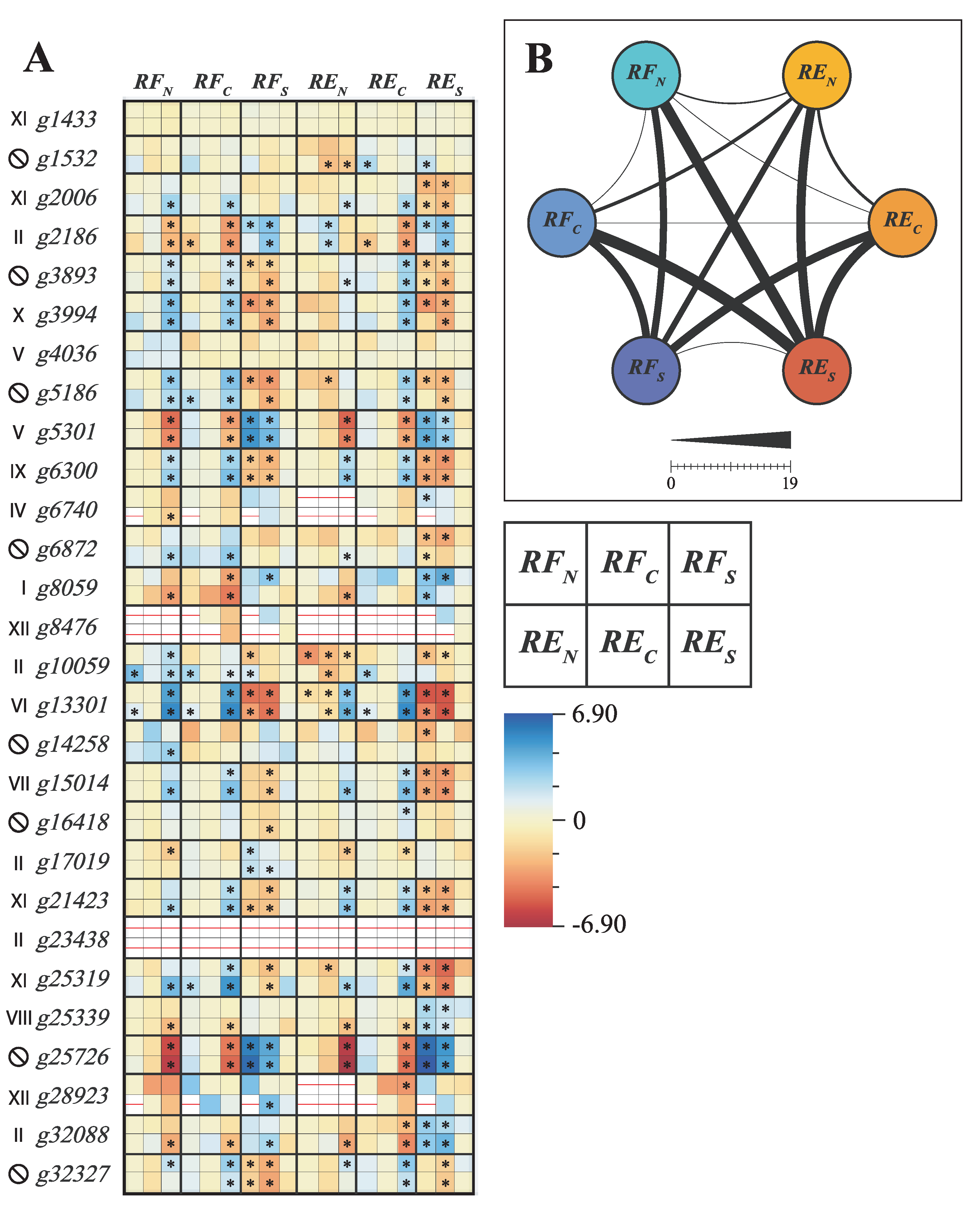
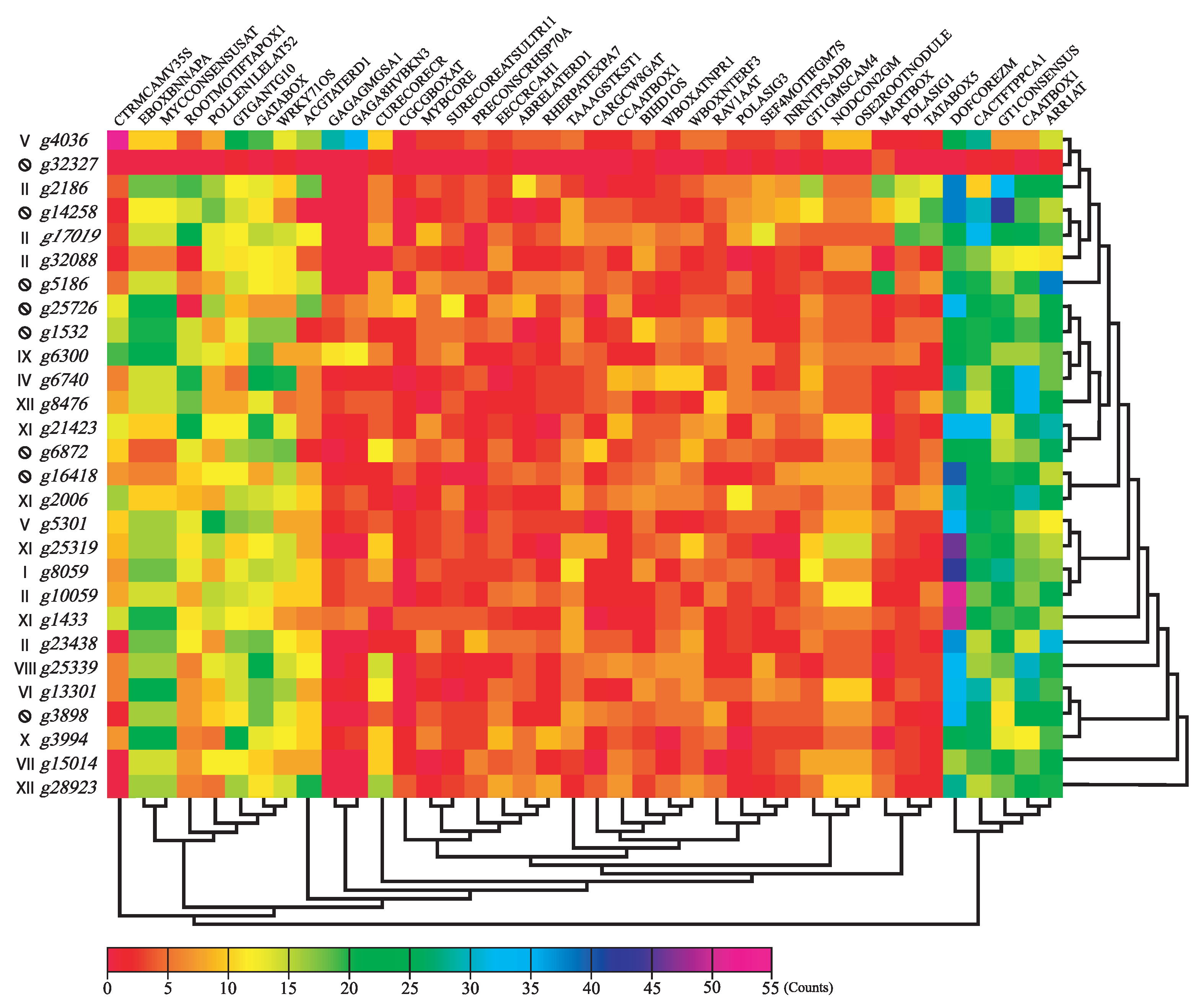
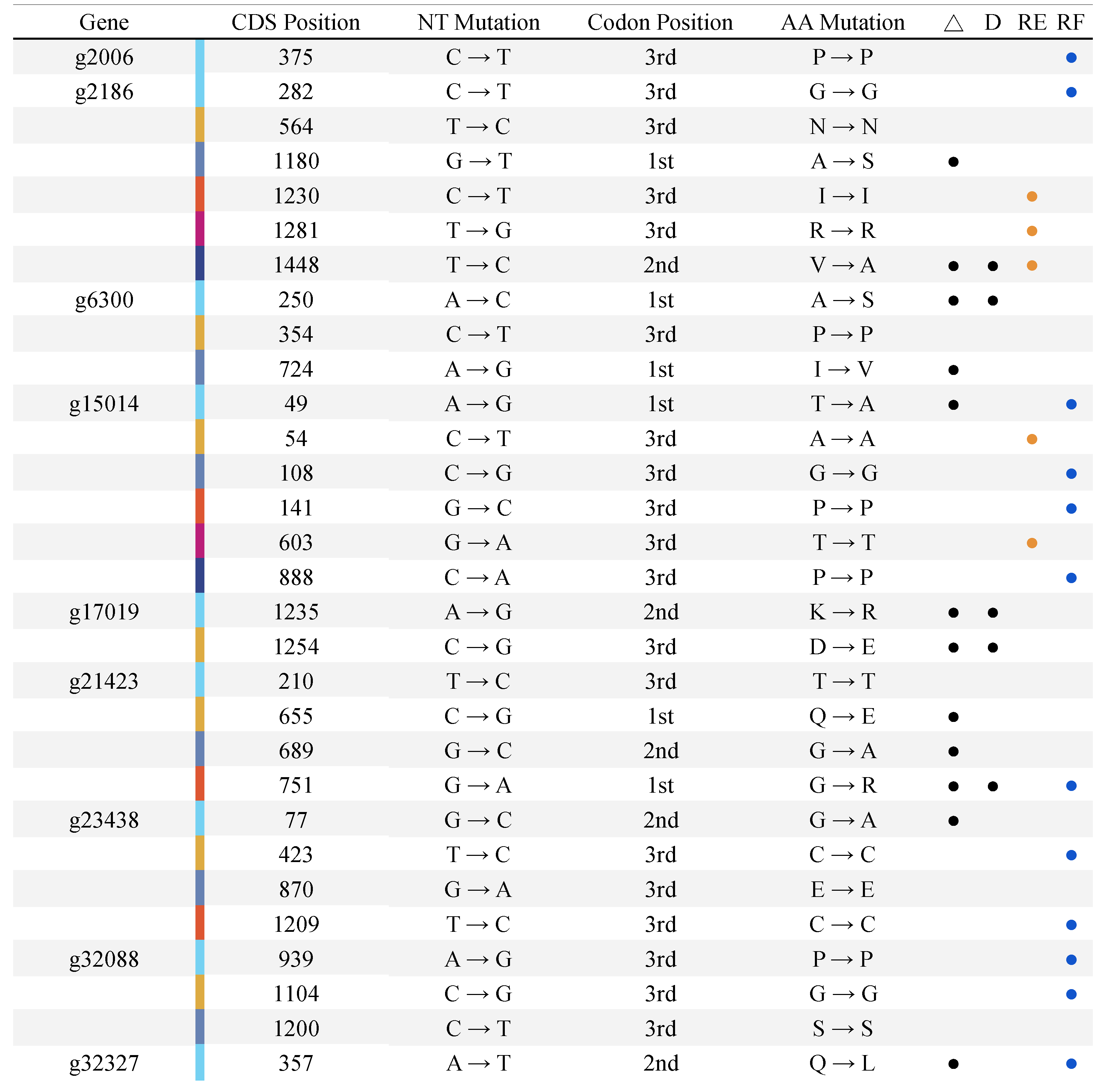
2.4. Differential Expression Analysis, SNP Indentification and Promoter Motif Enrichment Analysis
3. Results
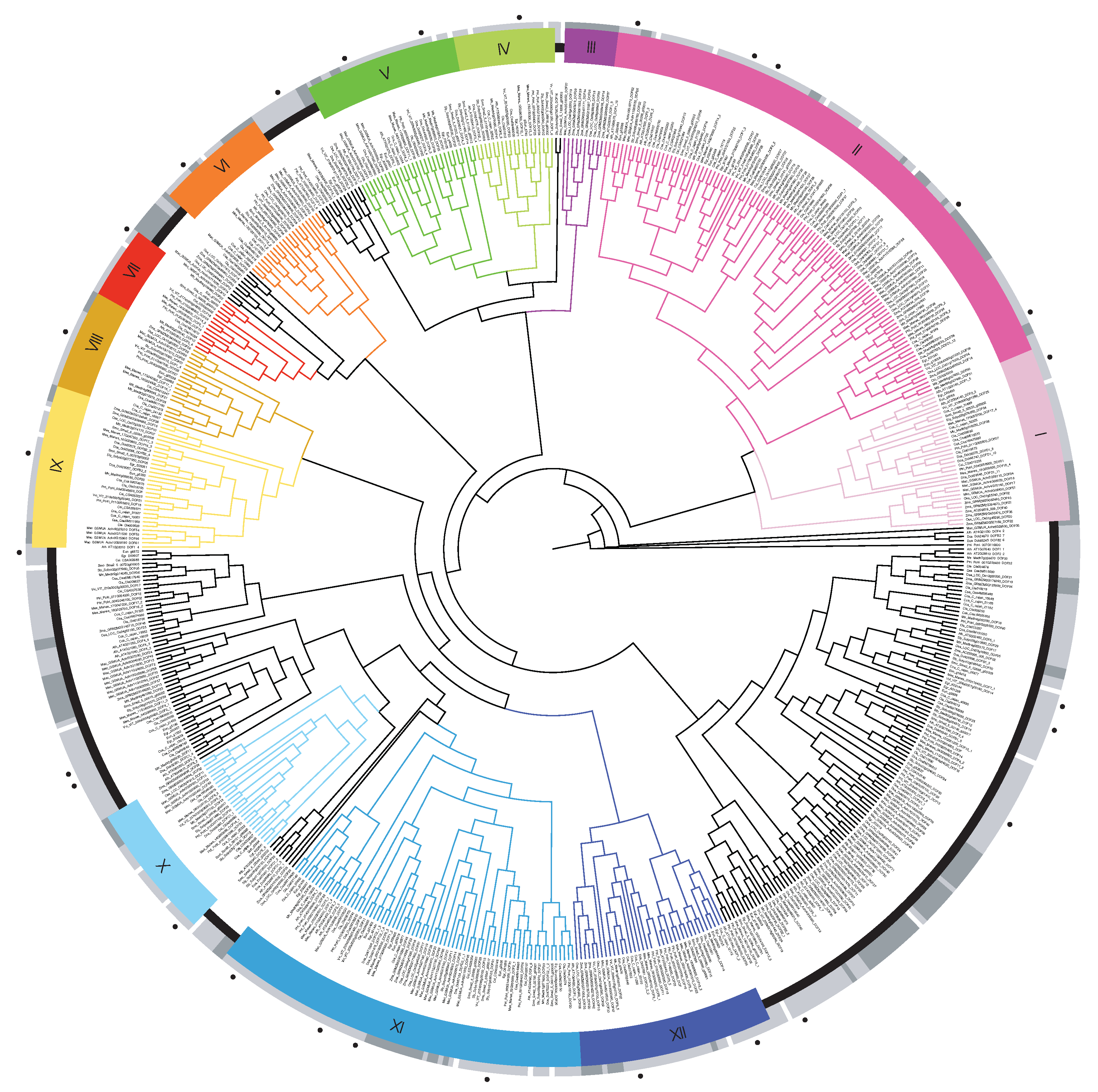
3.1. Identification and Classification of E. uniflora DOF Genes
3.2. Gene Structure and Domain Conservation of E. uniflora DOF Genes
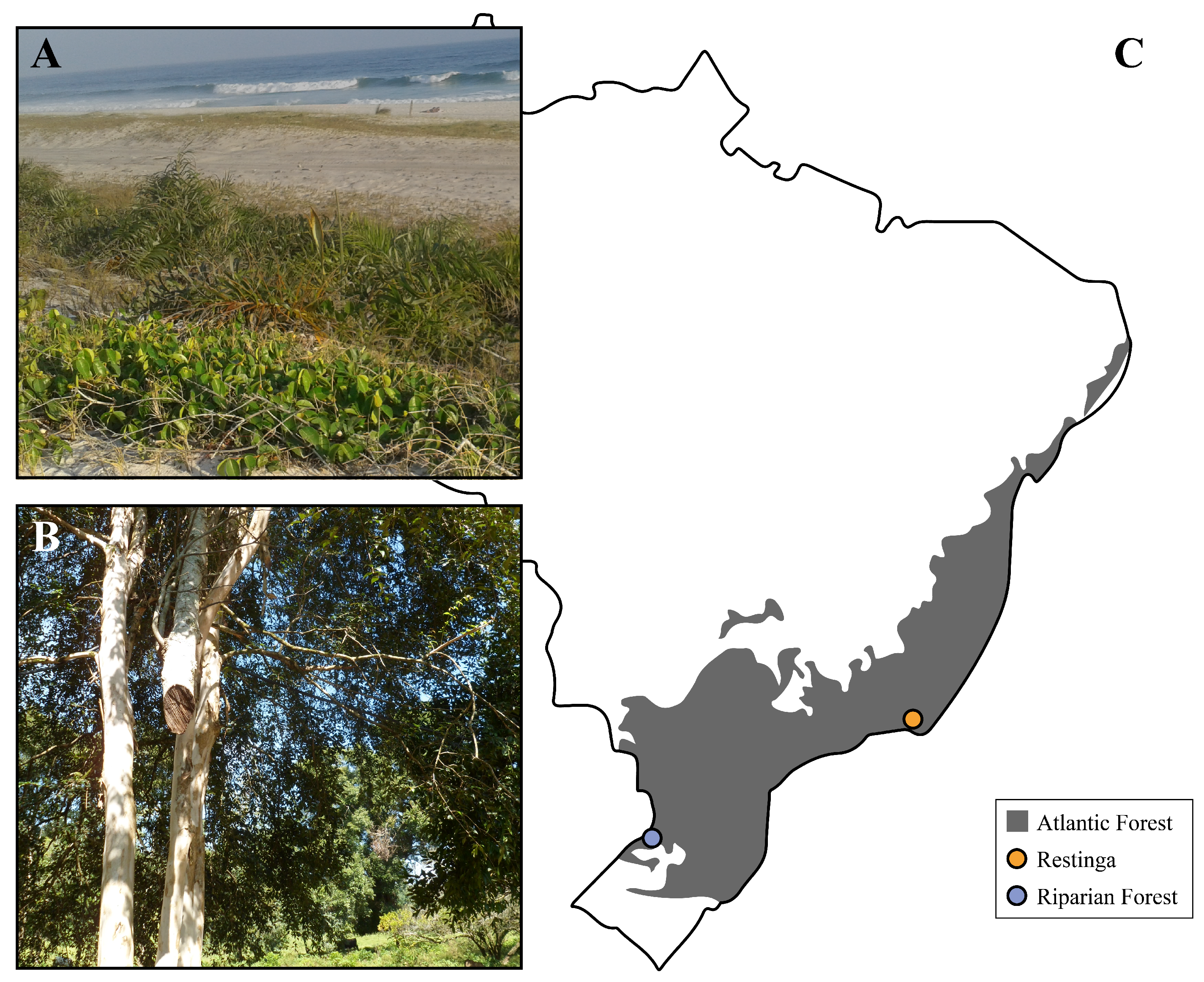
3.3. E. uniflora Populations and Their DOF Gene Arsenals
3.4. Promoter Region Analysis
4. Discussion
4.1. Phylogenetic Relationships Reveal DOF Genes Acting in Similar Pathways
4.2. DOF Genes as Drivers for Local Adaptation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Amino-acid |
| AF | Atlantic Forest |
| CDS | Coding sequence |
| DEG | Differentialy expressed genes |
| DOF | DNA-binding with one-finger |
| CDF | Cycling-dof-factor |
| FC | Fold-change |
| PP | Posterior probability |
| RE | Restinga |
| RF | Riparian Forest |
| TF | Transcription Factor |
| MCOG | Majorly Conserved Orthologous Group |
References
- Scarano, F. Biomas Brasileiros: Retrato de um País Plural; Casa da Palavra: Rio de Janeiro, Brazil, 2012. [Google Scholar]
- Lucas, E.J.; Bünger, M.O. Myrtaceae in the Atlantic forest: Their role as a ‘model’ group. Biodivers. Conserv. 2015, 24, 2165–2180. [Google Scholar] [CrossRef]
- Anton, D.B.; Guzman, F.L.; Vetö, N.M.; Krause, F.A.; Kulcheski, F.R.; Coelho, A.P.D.; Duarte, G.L.; Margis, R.; Dillenburg, L.R.; Turchetto-Zolet, A.C. Characterization and expression analysis of P5CS (Δ1-pyrroline-5-carboxylate synthase) gene in two distinct populations of the Atlantic Forest native species Eugenia uniflora L. Mol. Biol. Rep. 2020, 47, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- De Souza Neto, J.D.; Dos Santos, E.K.; Lucas, E.; Vetö, N.M.; Barrientos-Diaz, O.; Staggemeier, V.G.; Vasconcelos, T.; Turchetto-Zolet, A.C. Advances and perspectives on the evolutionary history and diversification of Neotropical Myrteae (Myrtaceae). Bot. J. Linn. Soc. 2022, 199, 173–195. [Google Scholar] [CrossRef]
- Falcão, T.R.; de Araújo, A.A.; Soares, L.A.L.; de Moraes Ramos, R.T.; Bezerra, I.C.F.; Ferreira, M.R.A.; de Souza Neto, M.A.; Melo, M.C.N.; de Araújo, R.F.; de Aguiar Guerra, A.C.V.; et al. Crude extract and fractions from Eugenia uniflora Linn leaves showed anti-inflammatory, antioxidant, and antibacterial activities. BMC Complement. Altern. Med. 2018, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Silva-Rocha, W.P.; de Brito Lemos, V.L.; Ferreira, M.R.A.; Soares, L.A.L.; Svidzisnki, T.I.E.; Milan, E.P.; Chaves, G.M. Effect of the crude extract of Eugenia uniflora in morphogenesis and secretion of hydrolytic enzymes in Candida albicans from the oral cavity of kidney transplant recipients. BMC Complement. Altern. Med. 2015, 15, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, K.K.A.; Matias, E.F.F.; Tintino, S.R.; Souza, C.E.S.; Braga, M.F.B.M.; Guedes, G.M.M.; Rolón, M.; Vega, C.; de Arias, A.R.; Costa, J.G.M.; et al. Anti-Trypanosoma cruzi and cytotoxic activities of Eugenia uniflora L. Exp. Parasitol. 2012, 131, 130–132. [Google Scholar] [CrossRef] [Green Version]
- Rattmann, Y.D.; de Souza, L.M.; Malquevicz-Paiva, S.M.; Dartora, N.; Sassaki, G.L.; Gorin, P.A.J.; Iacomini, M. Analysis of Flavonoids from Eugenia uniflora Leaves and Its Protective Effect against Murine Sepsis. Evid. Based Complement. Altern. Med. 2012, 2012, 623940. [Google Scholar] [CrossRef] [Green Version]
- Sobeh, M.; El-Raey, M.; Rezq, S.; Abdelfattah, M.; Petruk, G.; Osman, S.; El-Shazly, A.; El-Beshbishy, H.; Mahmoud, M.; Wink, M. Chemical profiling of secondary metabolites of Eugenia uniflora and their antioxidant, anti-inflammatory, pain killing and anti-diabetic activities: A comprehensive approach. J. Ethnopharmacol. 2019, 240, 111939. [Google Scholar] [CrossRef]
- Vetö, N.M.; Postolache, D.; Guzman Escudero, F.L.; Vajana, E.; Burgo Braga, R.; Salgueiro, F.; Margis, R.; Vendramin, G.G.; Turchetto-Zolet, A.C. Population structure and signals of local adaptation in Eugenia uniflora (Myrtaceae), a widely distributed species in the Atlantic Forest. Bot. J. Linn. Soc. 2022, boac012. [Google Scholar] [CrossRef]
- Turchetto-Zolet, A.C.; Salgueiro, F.; Turchetto, C.; Cruz, F.; Veto, N.M.; Barros, M.J.F.; Segatto, A.L.A.; Freitas, L.B.; Margis, R. Phylogeography and ecological niche modelling in Eugenia uniflora (Myrtaceae) suggest distinct vegetational responses to climate change between the southern and the northern Atlantic Forest. Bot. J. Linn. Soc. 2016, 182, 670–688. [Google Scholar] [CrossRef]
- Moreno-Risueno, M.Á.; Martínez, M.; Vicente-Carbajosa, J.; Carbonero, P. The family of DOF transcription factors: From green unicellular algae to vascular plants. Mol. Genet. Genom. 2007, 277, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lijavetzky, D.; Carbonero, P.; Vicente-Carbajosa, J. Genome-wide comparative phylogenetic analysis of the rice and Arabidopsis Dof gene families. BMC Evol. Biol. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Cao, J. Comparative Analysis of Dof Transcription Factor Family in Maize. Plant Mol. Biol. Report. 2015, 33, 1245–1258. [Google Scholar] [CrossRef]
- Kushwaha, H.; Gupta, S.; Singh, V.K.; Rastogi, S.; Yadav, D. Genome wide identification of Dof transcription factor gene family in sorghum and its comparative phylogenetic analysis with rice and Arabidopsis. Mol. Biol. Rep. 2011, 38, 5037–5053. [Google Scholar] [CrossRef]
- Yanagisawa, S. The Dof family of plant transcription factors. Trends Plant Sci. 2002, 7, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Zhang, K.; Chen, S.; Liu, M.; Zhang, Q. Genome-wide analysis and comparison of the DNA-binding one zinc finger gene family in diploid and tetraploid cotton (Gossypium). PLoS ONE 2020, 15, e0235317. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, Y.; Zhang, C.; Zhang, T.; Hu, T.; Ye, J.; Zhang, J.; Wang, T.; Li, H.; Ye, Z. Genome-wide Analysis of Plant-specific Dof Transcription Factor Family in Tomato. J. Integr. Plant Biol. 2013, 55, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Shigyo, M.; Tabei, N.; Yoneyama, T.; Yanagisawa, S. Evolutionary Processes During the Formation of the Plant-Specific Dof Transcription Factor Family. Plant Cell Physiol. 2007, 48, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, S.; Sanda, S.; Uraguchi, Y.; Nakagawa, S.; Sawayama, S. Overexpression of the DOF-Type Transcription Factor Enhances Lipid Synthesis in Chlorella vulgaris. Appl. Biochem. Biotechnol. 2019, 189, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Renau-Morata, B.; Carrillo, L.; Cebolla-Cornejo, J.; Molina, R.V.; Martí, R.; Domínguez-Figueroa, J.; Vicente-Carbajosa, J.; Medina, J.; Nebauer, S.G. The targeted overexpression of SlCDF4 in the fruit enhances tomato size and yield involving gibberellin signalling. Sci. Rep. 2020, 10, 10645. [Google Scholar] [CrossRef] [PubMed]
- Umemura, Y.; Ishiduka, T.; Yamamoto, R.; Esaka, M. The Dof domain, a zinc finger DNA-binding domain conserved only in higher plants, truly functions as a Cys2/Cys2 Zn finger domain. Plant J. 2004, 37, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, S.; Schmidt, R.J. Diversity and similarity among recognition sequences of Dof transcription factors. Plant J. 1999, 17, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; Panigrahi, K.C.; Gissot, L.; Sauerbrunn, N.; Rühl, M.; Jarillo, J.A.; Coupland, G. Arabidopsis DOF Transcription Factors Act Redundantly to Reduce CONSTANS Expression and Are Essential for a Photoperiodic Flowering Response. Dev. Cell 2009, 17, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Zhang, Y.; Zhang, W.; Gu, J.; Xiong, F.; An, G.; Wu, Y. Rice transcription factor OsDOF18 enlarges the starch granule size by cytokinin. Curr. Plant Biol. 2022, 31, 100253. [Google Scholar] [CrossRef]
- Qin, H.; Wang, J.; Chen, X.; Wang, F.; Peng, P.; Zhou, Y.; Miao, Y.; Zhang, Y.; Gao, Y.; Qi, Y.; et al. Rice OsDOF15 contributes to ethylene-inhibited primary root elongation under salt stress. New Phytol. 2019, 223, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, L.; Liu, X.; Chen, X.; Wang, X. Evolution analysis of Dof transcription factor family and their expression in response to multiple abiotic stresses in Malus domestica. Gene 2018, 639, 137–148. [Google Scholar] [CrossRef]
- Chawade, A.; Bräutigam, M.; Lindlöf, A.; Olsson, O.; Olsson, B. Putative cold acclimation pathways in Arabidopsis thaliana identified by a combined analysis of mRNA co-expression patterns, promoter motifs and transcription factors. BMC Genom. 2007, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Cooper, B.; Clarke, J.D.; Budworth, P.; Kreps, J.; Hutchison, D.; Park, S.; Guimil, S.; Dunn, M.; Luginbühl, P.; Ellero, C.; et al. A network of rice genes associated with stress response and seed development. Proc. Natl. Acad. Sci. USA 2003, 100, 4945–4950. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Neuman, D.; Shen, Q.J. Interactions of two transcriptional repressors and two transcriptional activators in modulating gibberellin signaling in aleurone cells. Plant Physiol. 2008, 148, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Shim, Y.; Kang, K.; An, G.; Paek, N.C. Rice DNA-Binding One Zinc Finger 24 (OsDOF24) Delays Leaf Senescence in a Jasmonate-Mediated Pathway. Plant Cell Physiol. 2019, 60, 2065–2076. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Briefings Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Malviya, N.; Gupta, S.; Singh, V.K.; Yadav, M.K.; Bisht, N.C.; Sarangi, B.K.; Yadav, D. Genome wide in silico characterization of Dof gene families of pigeonpea (Cajanus cajan (L) Millsp.). Mol. Biol. Rep. 2015, 42, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Li, C.; Zhang, J.; Tian, Y.; Wang, H.; Zhang, Y.; Zhang, Z.; Xiang, Q.; Han, X.; Zhang, L. Genome-wide identification and expression analysis of the Dof gene family under drought stress in tea (Camellia sinensis). PeerJ 2020, 8, e9269. [Google Scholar] [CrossRef]
- Zhou, Y.; Cheng, Y.; Wan, C.; Li, J.; Yang, Y.; Chen, J. Genome-wide characterization and expression analysis of the Dof gene family related to abiotic stress in watermelon. PeerJ 2020, 8, e8358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.l.; Cheng, Q.; Zhao, L.; Mao, A.; Yang, J.; Yu, S.; Weng, Y.; Xu, Y. Identification and characterisation of Dof transcription factors in the cucumber genome. Sci. Rep. 2016, 6, 23072. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Huang, Y.; Li, M.y.; Wang, F.; Xu, Z.s.; Xiong, A.s. Dof transcription factors in carrot: Genome-wide analysis and their response to abiotic stress. Biotechnol. Lett. 2016, 38, 145–155. [Google Scholar] [CrossRef] [PubMed]
- d’Almeida, G.S.; Breton, M.; Camargo, S.; Frazzon, J.; Pasquali, G. Phylogenetic comparative and expression analysis of genes encoding dof transcription factors from Eucalyptus grandis. BMC Proc. 2011, 5, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Zhu, J.; Zhang, X. Genome-wide identification and characterization of the Dof gene family in cassava (Manihot esculenta). Gene 2019, 687, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.J.; Song, L.L.; Zhang, J.; Liu, Y.; Guo, C.H. Genome-wide identification and characterization of the Dof gene family in Medicago truncatula. Genet. Mol. Res. 2015, 14, 10645–10657. [Google Scholar] [CrossRef]
- Dong, C.; Hu, H.; Xie, J. Genome-wide analysis of the DNA-binding with one zinc finger (Dof) transcription factor family in bananas. Genome 2016, 59, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, S.; Gao, Y.; Yang, J. Characterization of Dof Transcription Factors and Their Responses to Osmotic Stress in Poplar (Populus trichocarpa). PLoS ONE 2017, 12, e0170210. [Google Scholar] [CrossRef] [Green Version]
- da Silva, D.C.; da Silveira Falavigna, V.; Fasoli, M.; Buffon, V.; Porto, D.D.; Pappas, G.J.; Pezzotti, M.; Pasquali, G.; Revers, L.F. Transcriptome analyses of the Dof-like gene family in grapevine reveal its involvement in berry, flower and seed development. Hortic. Res. 2016, 3, 16042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Deeba, F.; Sultana, T.; Mahmood, T.; O’Shea, C.; Skriver, K.; Naqvi, S.M.S. Involvement of WRKY, MYB and DOF DNA-binding proteins in interaction with a rice germin-like protein gene promoter. Acta Physiol. Plant. 2017, 39, 189. [Google Scholar] [CrossRef]
- Lorrai, R.; Gandolfi, F.; Boccaccini, A.; Ruta, V.; Possenti, M.; Tramontano, A.; Costantino, P.; Lepore, R.; Vittorioso, P. Genome-wide RNA-seq analysis indicates that the DAG1 transcription factor promotes hypocotyl elongation acting on ABA, ethylene and auxin signaling. Sci. Rep. 2018, 8, 15895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondhare, K.R.; Vetal, P.V.; Kalsi, H.S.; Banerjee, A.K. BEL1-like protein (StBEL5) regulates CYCLING DOF FACTOR1 (StCDF1) through tandem TGAC core motifs in potato. J. Plant Physiol. 2019, 241, 153014. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Malviya, N.; Kushwaha, H.; Nasim, J.; Bisht, N.C.; Singh, V.K.; Yadav, D. Insights into structural and functional diversity of Dof (DNA binding with one finger) transcription factor. Planta 2015, 241, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ma, Y.; Lu, Y.; Yue, J.; Ming, R. Expression profiling of the Dof gene family under abiotic stresses in spinach. Sci. Rep. 2021, 11, 14429. [Google Scholar] [CrossRef]
- Hamdi, J.; Kmeli, N.; Bouktila, D. Genome-wide survey of sugar beet (Beta vulgaris subsp. vulgaris) Dof transcription factors reveals structural diversity, evolutionary expansion and involvement in taproot development and biotic stress response. Biologia 2021, 76, 2421–2436. [Google Scholar] [CrossRef]
- Noguero, M.; Atif, R.M.; Ochatt, S.; Thompson, R.D. The role of the DNA-binding One Zinc Finger (DOF) transcription factor family in plants. Plant Sci. 2013, 209, 32–45. [Google Scholar] [CrossRef]
- Renau-Morata, B.; Carrillo, L.; Dominguez-Figueroa, J.; Vicente-Carbajosa, J.; Molina, R.V.; Nebauer, S.G.; Medina, J. CDF transcription factors: Plant regulators to deal with extreme environmental conditions. J. Exp. Bot. 2020, 71, 3803–3815. [Google Scholar] [CrossRef]
- Corrales, A.R.; Nebauer, S.G.; Carrillo, L.; Fernández-Nohales, P.; Marqués, J.; Renau-Morata, B.; Granell, A.; Pollmann, S.; Vicente-Carbajosa, J.; Molina, R.V.; et al. Characterization of tomato Cycling Dof Factors reveals conserved and new functions in the control of flowering time and abiotic stress responses. J. Exp. Bot. 2014, 65, 995–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaton, D.D.; Smith, R.W.; Song, Y.H.; MacGregor, D.R.; Stewart, K.; Steel, G.; Foreman, J.; Penfield, S.; Imaizumi, T.; Millar, A.J.; et al. Linked circadian outputs control elongation growth and flowering in response to photoperiod and temperature. Mol. Syst. Biol. 2015, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Schultz, T.F.; Harmon, F.G.; Ho, L.A.; Kay, S.A. FKF1 F-Box Protein Mediates Cyclic Degradation of a Repressor of CONSTANS in Arabidopsis. Science 2005, 309, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Goralogia, G.S.; Liu, T.K.; Zhao, L.; Panipinto, P.M.; Groover, E.D.; Bains, Y.S.; Imaizumi, T. CYCLING DOF FACTOR 1 represses transcription through the TOPLESS co-repressor to control photoperiodic flowering in Arabidopsis. Plant J. 2017, 92, 244–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloosterman, B.; Abelenda, J.A.; Gomez, M.d.M.C.; Oortwijn, M.; de Boer, J.M.; Kowitwanich, K.; Horvath, B.M.; van Eck, H.J.; Smaczniak, C.; Prat, S.; et al. Naturally occurring allele diversity allows potato cultivation in northern latitudes. Nature 2013, 495, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Bueso, E.; Muñoz-Bertomeu, J.; Campos, F.; Martínez, C.; Tello, C.; Martínez-Almonacid, I.; Ballester, P.; Simón-Moya, M.; Brunaud, V.; Yenush, L.; et al. Arabidopsis COGWHEEL1 links light perception and gibberellins with seed tolerance to deterioration. Plant J. 2016, 87, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.H.; Lim, P.O.; Kim, J.S.; Cho, D.S.; Hong, S.H.; Nam, H.G. The Arabidopsis COG1 gene encodes a Dof domain transcription factor and negatively regulates phytochrome signaling. Plant J. 2003, 34, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Chen, H.; Cai, W. Transcription factor CDF4 promotes leaf senescence and floral organ abscission by regulating abscisic acid and reactive oxygen species pathways in Arabidopsis. EMBO Rep. 2020, 21, e48967. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Yuan, T.; Tarkowská, D.; Kim, J.; Nam, H.G.; Novák, O.; He, K.; Gou, X.; Li, J. Brassinosteroid Biosynthesis Is Modulated via a Transcription Factor Cascade of COG1, PIF4, and PIF5. Plant Physiol. 2017, 174, 1260–1273. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Liu, X.; Yin, D.; Yuan, H.; Xie, Q.; Zhao, X.; Li, X.; Zhu, L.; Li, S.; Li, D. Constitutive expression of OsDof4, encoding a C2-C2 zinc finger transcription factor, confesses its distinct flowering effects under long- and short-day photoperiods in rice (Oryza sativa L.). BMC Plant Biol. 2017, 17, 166. [Google Scholar] [CrossRef] [Green Version]
- Deepika; Singh, A. Expression dynamics indicate the role of Jasmonic acid biosynthesis pathway in regulating macronutrient (N, P and K+) deficiency tolerance in rice (Oryza sativa L.). Plant Cell Rep. 2021, 40, 1495–1512. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Yanagisawa, S. Transcriptional repression caused by Dof5.8 is involved in proper vein network formation in Arabidopsis thaliana leaves. J. Plant Res. 2015, 128, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Donner, T.J.; Scarpella, E.; Yanagisawa, S. MONOPTEROS directly activates the auxin-inducible promoter of the Dof5.8 transcription factor gene in Arabidopsis thaliana leaf provascular cells. J. Exp. Bot. 2015, 66, 283–291. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Su, C.; Wang, Y.; Wei, Z. ATDOF5.8 protein is the upstream regulator of ANAC069 and is responsive to abiotic stress. Biochimie 2015, 110, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.E.; McGregor, S.R.; Sun, H.; Gough, C.; Bågman, A.M.; Soyars, C.L.; Kroon, J.T.; Gaudinier, A.; Williams, C.J.; Yang, X.; et al. A PXY-Mediated Transcriptional Network Integrates Signaling Mechanisms to Control Vascular Development in Arabidopsis. Plant Cell 2020, 32, 319–335. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Gong, Q.; Bohnert, H.J. Dissecting salt stress pathways. J. Exp. Bot. 2006, 57, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Thieme, C.J.; Rojas-Triana, M.; Stecyk, E.; Schudoma, C.; Zhang, W.; Yang, L.; Miñambres, M.; Walther, D.; Schulze, W.X.; Paz-Ares, J.; et al. Endogenous Arabidopsis messenger RNAs transported to distant tissues. Nat. Plants 2015, 1, 15025. [Google Scholar] [CrossRef]
- Xu, P.; Chen, H.; Ying, L.; Cai, W. AtDOF5.4/OBP4, a DOF Transcription Factor Gene that Negatively Regulates Cell Cycle Progression and Cell Expansion in Arabidopsis thaliana. Sci. Rep. 2016, 6, 27705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Qin, G.; Gu, H.; Qu, L.J. Dof5.6/HCA2, a Dof Transcription Factor Gene, Regulates Interfascicular Cambium Formation and Vascular Tissue Development in Arabidopsis. Plant Cell 2009, 21, 3518–3534. [Google Scholar] [CrossRef] [Green Version]
- Gualberti, G.; Papi, M.; Bellucci, L.; Ricci, I.; Bouchez, D.; Camilleri, C.; Costantino, P.; Vittorioso, P. Mutations in the Dof Zinc Finger Genes DAG2 and DAG1 Influence with Opposite Effects the Germination of Arabidopsis Seeds. Plant Cell 2002, 14, 1253–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.; Yamaguchi, S.; Hu, J.; Yusuke, J.; Jung, B.; Paik, I.; Lee, H.S.; Sun, T.P.; Kamiya, Y.; Choi, G. PIL5, a Phytochrome-Interacting bHLH Protein, Regulates Gibberellin Responsiveness by Binding Directly to the GAI and RGA Promoters in Arabidopsis Seeds. Plant Cell 2007, 19, 1192–1208. [Google Scholar] [CrossRef] [Green Version]
- Piskurewicz, U.; Turečková, V.; Lacombe, E.; Lopez-Molina, L. Far-red light inhibits germination through DELLA-dependent stimulation of ABA synthesis and ABI3 activity. EMBO J. 2009, 28, 2259–2271. [Google Scholar] [CrossRef] [Green Version]
- Gabriele, S.; Rizza, A.; Martone, J.; Circelli, P.; Costantino, P.; Vittorioso, P. The Dof protein DAG1 mediates PIL5 activity on seed germination by negatively regulating GA biosynthetic gene AtGA3ox1: DAG1 represses seed germination via PIL5 signalling. Plant J. 2009, 61, 312–323. [Google Scholar] [CrossRef]
- Boccaccini, A.; Santopolo, S.; Capauto, D.; Lorrai, R.; Minutello, E.; Serino, G.; Costantino, P.; Vittorioso, P. The DOF protein DAG1 and the DELLA protein GAI cooperate in negatively regulating the AtGA3ox1 gene. Mol. Plant 2014, 7, 1486–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccaccini, A.; Lorrai, R.; Ruta, V.; Frey, A.; Mercey-Boutet, S.; Marion-Poll, A.; Tarkowská, D.; Strnad, M.; Costantino, P.; Vittorioso, P. The DAG1 transcription factor negatively regulates the seed-to-seedling transition in Arabidopsis acting on ABA and GA levels. BMC Plant Biol. 2016, 16, 198. [Google Scholar] [CrossRef] [Green Version]
- Santopolo, S.; Boccaccini, A.; Lorrai, R.; Ruta, V.; Capauto, D.; Minutello, E.; Serino, G.; Costantino, P.; Vittorioso, P. DOF AFFECTING GERMINATION 2 is a positive regulator of light-mediated seed germination and is repressed by DOF AFFECTING GERMINATION 1. BMC Plant Biol. 2015, 15, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ahmad, M.; Rim, Y.; Lucas, W.J.; Kim, J. Evolutionary and molecular analysis of Dof transcription factors identified a conserved motif for intercellular protein trafficking. New Phytol. 2013, 198, 1250–1260. [Google Scholar] [CrossRef]
- Miyashima, S.; Roszak, P.; Sevilem, I.; Toyokura, K.; Blob, B.; Heo, J.o.; Mellor, N.; Help-Rinta-Rahko, H.; Otero, S.; Smet, W.; et al. Mobile PEAR transcription factors integrate positional cues to prime cambial growth. Nature 2019, 565, 490–494. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Lenght (Aa) | Weight (kDa) | pI | Subcellular Location |
|---|---|---|---|---|
| g1433 | 276 | 29,822 | 8.85 | Nucleus |
| g1532 | 331 | 36,445 | 6.52 | Nucleus |
| g2006 | 307 | 33,047 | 8.79 | Nucleus |
| g2186 | 507 | 55,049 | 6.10 | Nucleus |
| g3898 | 332 | 35,571 | 9.19 | Nucleus |
| g3994 | 340 | 36,687 | 6.72 | Nucleus |
| g4036 | 193 | 21,308 | 7.55 | Nucleus |
| g5186 | 306 | 32,971 | 6.74 | Nucleus |
| g5301 | 243 | 26,111 | 8.54 | Nucleus |
| g6300 | 381 | 41,256 | 8.70 | Nucleus |
| g6740 | 259 | 26,047 | 8.45 | Nucleus |
| g6872 | 268 | 28,331 | 9.38 | Nucleus |
| g8059 | 186 | 20,727 | 9.62 | Nucleus |
| g8476 | 295 | 31,875 | 5.87 | Nucleus |
| g10059 | 491 | 52,622 | 8.37 | Nucleus |
| g13301 | 360 | 38,370 | 9.07 | Nucleus |
| g14258 | 370 | 38,939 | 8.82 | Nucleus |
| g15014 | 325 | 34,705 | 6.51 | Nucleus |
| g16418 | 362 | 38,399 | 9.59 | Nucleus |
| g17019 | 458 | 49,638 | 6.87 | Nucleus |
| g21423 | 258 | 27,408 | 9.55 | Nucleus |
| g23438 | 467 | 50,378 | 5.23 | Nucleus |
| g25319 | 308 | 32,858 | 8.50 | Nucleus |
| g25339 | 303 | 33,578 | 8.45 | Nucleus |
| g25726 | 253 | 25,782 | 9.13 | Nucleus |
| g28923 | 269 | 29,061 | 5.05 | Nucleus |
| g32088 | 447 | 48,074 | 8.89 | Nucleus |
| g32327 | 291 | 31,451 | 9.76 | Nucleus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waschburger, E.L.; Guzman, F.; Turchetto-Zolet, A.C. Genome-Wide Identification and Analysis of DOF Gene Family in Eugenia uniflora L. (Myrtaceae). Genes 2022, 13, 2235. https://doi.org/10.3390/genes13122235
Waschburger EL, Guzman F, Turchetto-Zolet AC. Genome-Wide Identification and Analysis of DOF Gene Family in Eugenia uniflora L. (Myrtaceae). Genes. 2022; 13(12):2235. https://doi.org/10.3390/genes13122235
Chicago/Turabian StyleWaschburger, Edgar Luis, Frank Guzman, and Andreia Carina Turchetto-Zolet. 2022. "Genome-Wide Identification and Analysis of DOF Gene Family in Eugenia uniflora L. (Myrtaceae)" Genes 13, no. 12: 2235. https://doi.org/10.3390/genes13122235