

Transcriptome Level Reveals the Triterpenoid Saponin Biosynthesis Pathway of Bupleurum falcatum L.

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling of B. falcatum
2.2. Saikosaponin Extraction and HPLC (High-Performance Liquid Chromatography)
2.3. PacBio Iso-Seq Library Construction and Transcriptome Analyses
2.4. PacBio Sequencing Read Processing
2.5. Functional Annotation of Genes
2.6. TF Prediction and lncRNA, SSR Analysis
2.7. Quantification of Gene Expression and Differentially Expressed Genes (DEGs) Analysis
2.8. Selecting Candidate Genes of Saikosaponin Biosynthesis
2.9. WGCNA
2.10. Validation by qRT-PCR
3. Results
3.1. Saikosaponin a (SSa) and Saikosaponins d (SSd) Content
3.2. Transcriptome Profile Analysis of B. falcatum
3.3. Functional Annotation of Genes
3.4. Ts Prediction and SSR: LncRNA Analysis
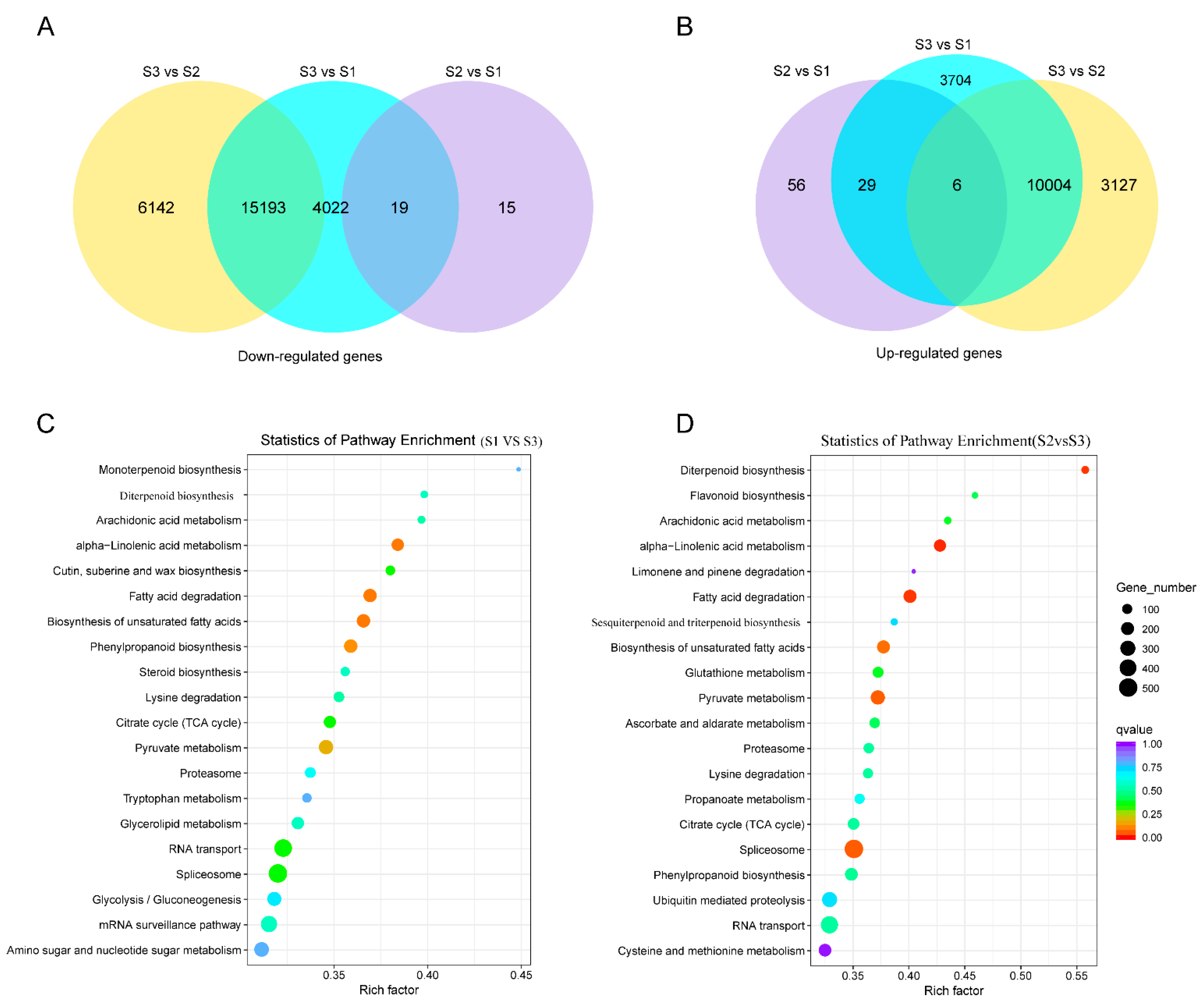
3.5. Comparative Analysis of DEGs
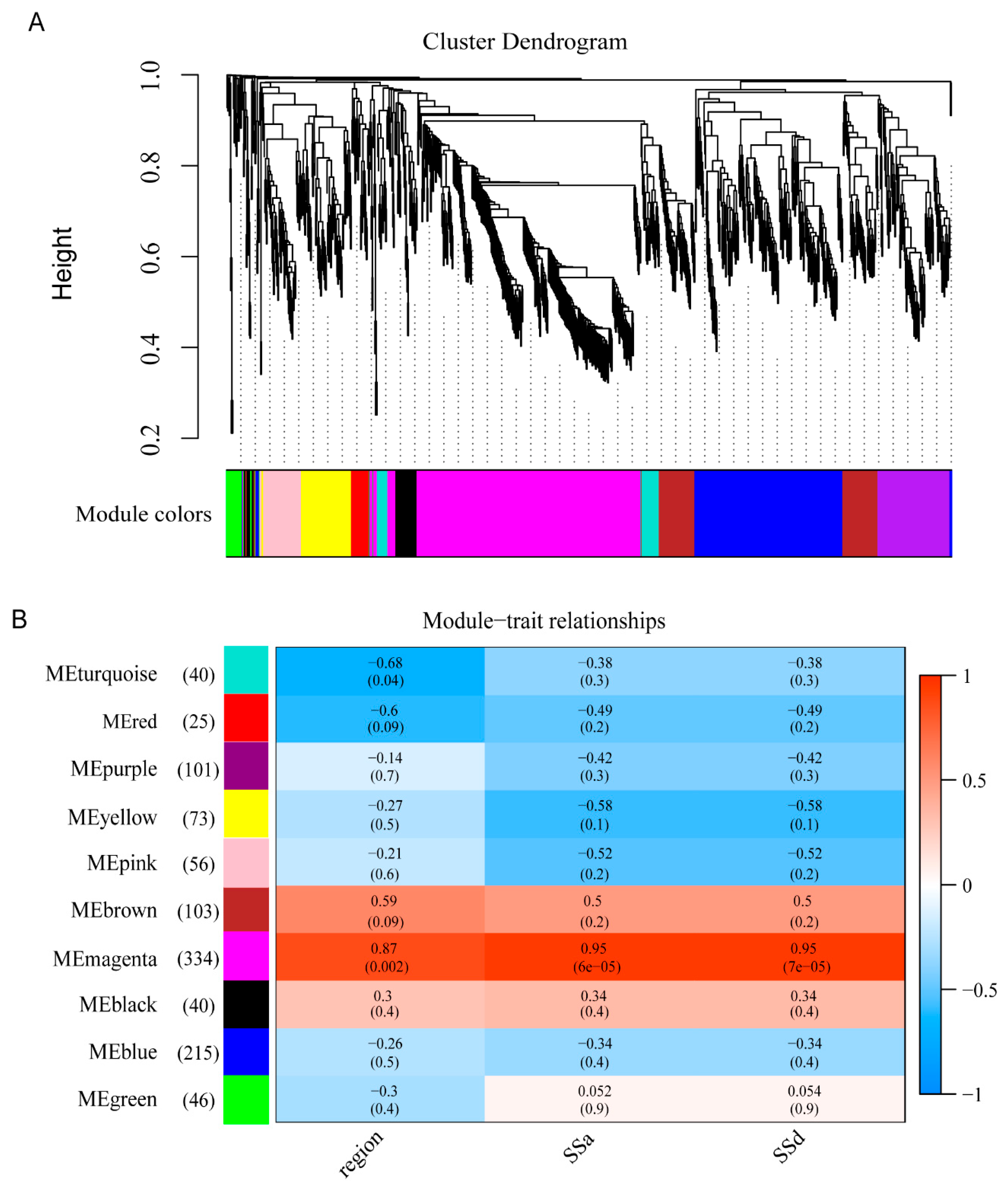
3.6. Gene Co-Expression Network Construction
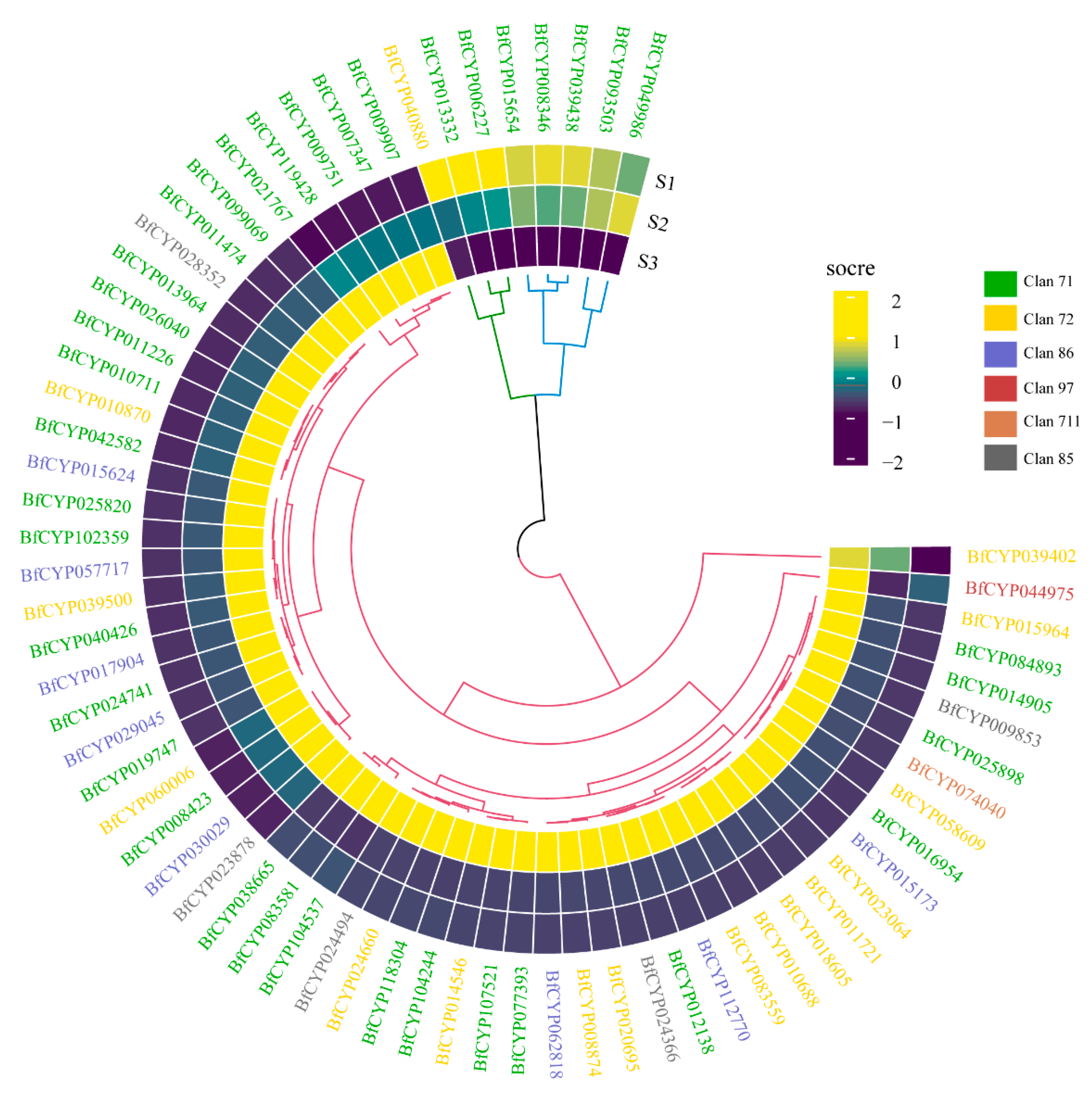
3.7. DEGs in MEmagenta Module Involved in the Saikosaponin Biosynthesis
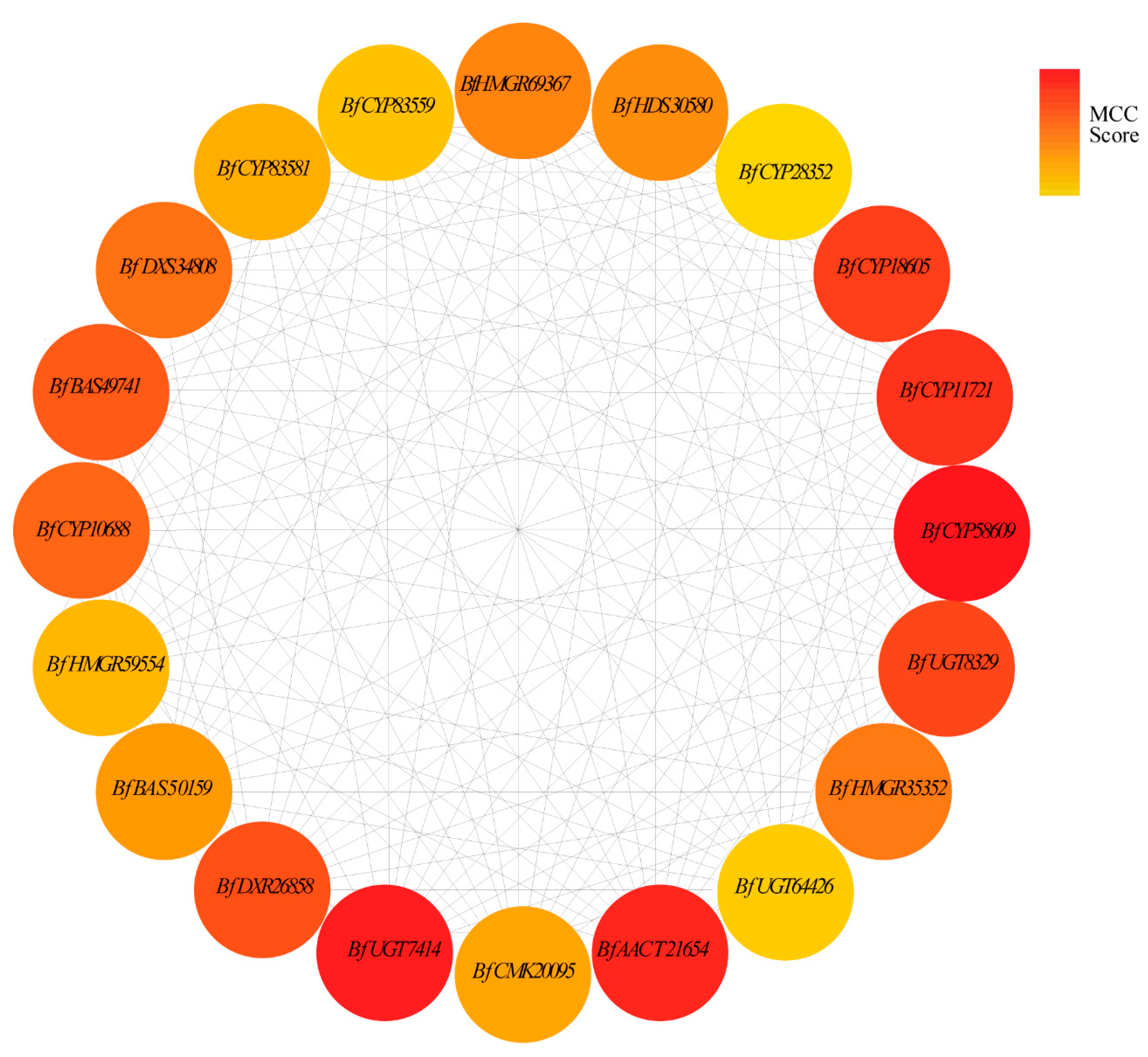
3.8. Hub Gene Selection for the MEmagenta Module
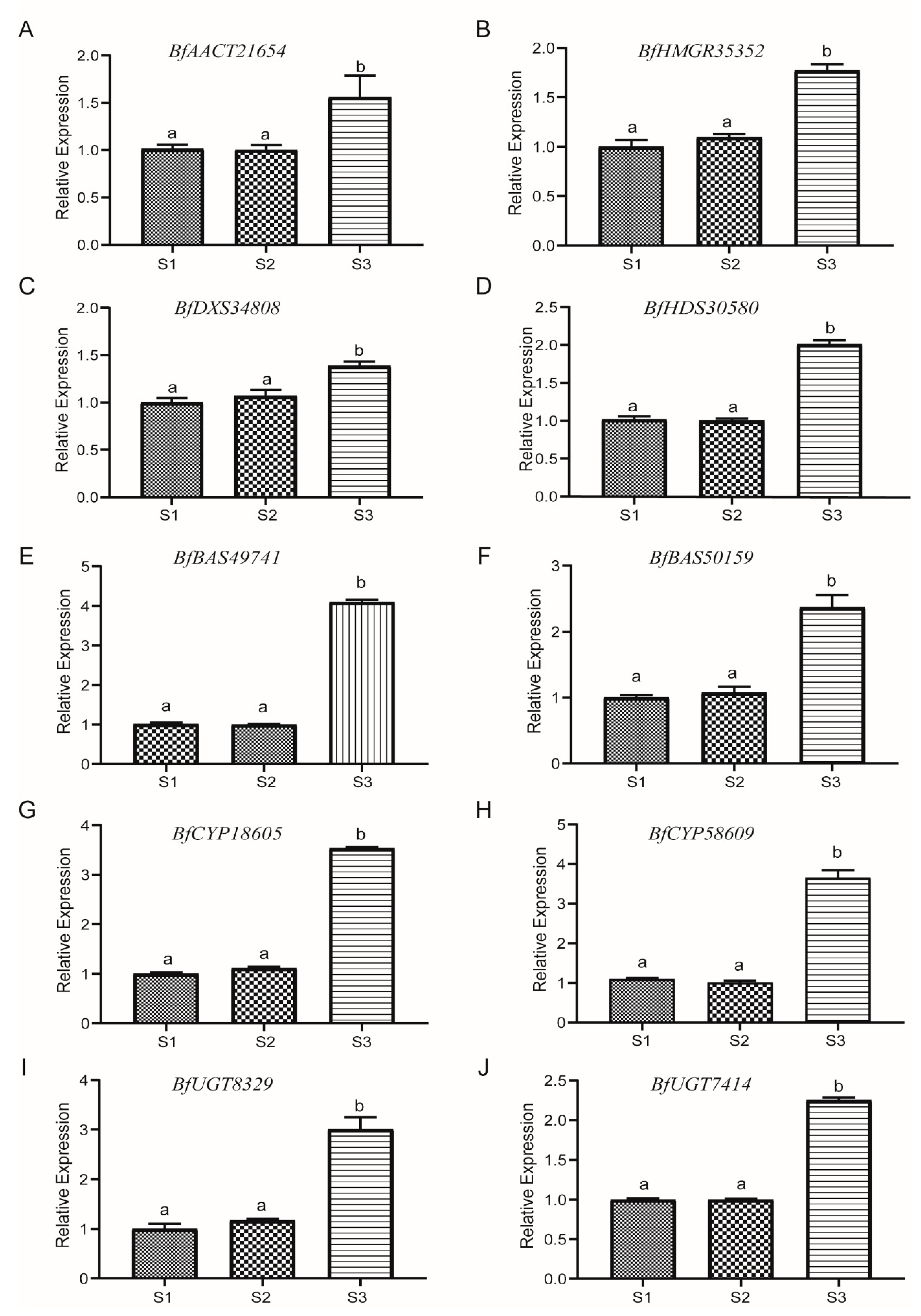
3.9. qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ashour, M.L.; Wink, M. Genus Bupleurum: A review of its phytochemistry, pharmacology and modes of action. J. Pharm. Pharm. 2011, 63, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.; Han, W.J.; Zhu, C.R.; Wei, J.H. Recent Progress in Saikosaponin Biosynthesis in Bupleurum. Curr. Pharm. Biotechnol. 2021, 22, 329–340. [Google Scholar] [CrossRef]
- Li, X.Q.; Song, Y.N.; Wang, S.J.; Rahman, K.; Zhu, J.Y.; Zhang, H. Saikosaponins: A review of pharmacological effects. J. Asian Nat. Prod. Res. 2018, 20, 399–411. [Google Scholar] [CrossRef]
- Yuan, B.; Yang, R.; Ma, Y.; Zhou, S.; Zhang, X.; Liu, Y. A systematic review of the active saikosaponins and extracts isolated from Radix Bupleuri and their applications. Pharm. Biol. 2017, 55, 620–635. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Oh, K.; Wang, Y.; Yi, T.; Chen, H.; Zhao, Z. Cell type-specific qualitative and quantitative analysis of saikosaponins in three Bupleurum species using laser microdissection and liquid chromatography-quadrupole/time of flight-mass spectrometry. J. Pharm. Biomed. Anal. 2014, 97, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Dai, Y.; Liu, N.; Li, Z.; Liu, X.; Hu, J.; Choi, Y.; Verpoorte, R. Metabolic Fingerprinting by 1HNMR for Discrimination of the Two Species Used as Radix Bupleuri. Planta Med. 2012, 78, 926–933. [Google Scholar] [CrossRef]
- Yu, M.; Chen, H.; Liu, Q.; Huang, J.; Semagn, K.; Liu, D.; Li, Y.C.; Yang, B.; He, Y.L.; Sui, C.; et al. Analysis of unigenes involved in lateral root development in Bupleurum chinense and B. scorzonerifolium. Planta 2021, 253, 128–139. [Google Scholar] [CrossRef]
- Gong, J.; Liu, M.; Xu, S.; Jiang, Y.; Pan, Y.; Zhai, Z.; Luo, Q.; Yang, L.; Wang, Y. Effects of light deficiency on the accumulation of saikosaponins and the ecophysiological characteristics of wild Bupleurum chinense DC. in China. Ind. Crops Prod. 2017, 99, 179–188. [Google Scholar] [CrossRef]
- Sun, J.L.; Duan, Z.J.; Zhang, Y.; Cao, S.S.; Tang, Z.H.; Abozeid, A. Metabolite Profiles Provide Insights into Underlying Mechanism in Bupleurum (Apiaceae) in Response to Three Levels of Phosphorus Fertilization. Plants 2022, 11, 752. [Google Scholar] [CrossRef]
- Meng, J.; Chen, X.F.; Yang, W.Y.; Yao, R.Y.; Song, J.H.; Li, Z.F.; Yang, X.W. Determination of saikosaponin a and d in taproot and lateral root of Bupleurum chinense DC. Chin. J. Pharma. Anal. 2013, 33, 1218–1222. [Google Scholar]
- Shon, T.K.; Agung, T.; Haryanto, D.; Yoshida, T. Variation and Distribution and Saikosaponin in Bupleurum falcatum L. J. Fac. Agric. Kyushu Univ. 1997, 42, 17–22. [Google Scholar] [CrossRef]
- Zheng, Y.J.; Xia, P.G.; Zhao, H.G.; Zheng, J.F.; Chai, W.G.; Liang, Z.S.; Yan, K.J. Suitable soil moisture contents for water use efficiency and saponins accumulation in Panax Notoginseng. Chin. Herb. Med. 2021, 13, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Wang, G.; Wei, X.; Chen, S.; Han, J. How to improve CHMs quality: Enlighten from CHMs ecological cultivation. Chin. Herb. Med. 2021, 13, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.L.; Cai, X.; Zheng-Hai, H.U.; Ni, X.L. Localization and Dynamic Change of Saikosaponin in Root of Bupleurum chinense. J. Integr. Plant Biol. 2008, 50, 7. [Google Scholar] [CrossRef] [PubMed]
- Seki, H.; Tamura, K.; Muranaka, T. P450s and UGTs: Key Players in the Structural Diversity of Triterpenoid Saponins. Plant Cell Physiol. 2015, 56, 1463–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Xu, H.; Ma, X.; Zhan, R.; Chen, W. Triterpenoid saponin biosynthetic pathway profiling and candidate gene mining of the Ilex asprella root using RNA-Seq. Int. J. Mol. Sci. 2014, 15, 5970–5987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, T.; Dwivedi, U.N. Plant triterpenoid saponins: Biosynthesis, in vitro production, and pharmacological relevance. Protoplasma 2019, 256, 1463–1486. [Google Scholar] [CrossRef]
- Moses, T.; Pollier, J.; Almagro, L.; Buyst, D.; Van Montagu, M.; Pedreno, M.A.; Martins, J.C.; Thevelein, J.M.; Goossens, A. Combinatorial biosynthesis of sapogenins and saponins in Saccharomyces cerevisiae using a C-16alpha hydroxylase from Bupleurum falcatum. Proc. Natl. Acad. Sci. USA 2014, 111, 1634–1639. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.Y.; Peng, P.H.; Lin, T.Y. Cloning and characterization of β-amyrin synthase from Bupleurum kaoi. In Proceedings of the 8th International Congress of Plant Molecular Biology, Adelaide, Australia, 20–25 August 2005. [Google Scholar]
- Gao, K.; Xu, J.S.; Sun, J.; Xu, Y.H.; Wei, J.H.; Sui, C.; Development, I.O.M.P.; Sciences, C.A.O.M. Molecular Cloning and Expression of Squalene Epoxidase from a Medicinal Plant, Bupleurum chinense. Chin. Herb. Med. 2016, 8, 67–74. [Google Scholar] [CrossRef]
- Kim, Y.S.; Cho, J.H.; Park, S.; Han, J.Y.; Back, K.; Choi, Y.E. Gene regulation patterns in triterpene biosynthetic pathway driven by overexpression of squalene synthase and methyl jasmonate elicitation in Bupleurum falcatum. Planta 2011, 233, 343–355. [Google Scholar] [CrossRef]
- Yu, M.; Chen, H.; Liu, S.; Li, Y.; Sui, C.; Hou, D.; Wei, J. Differential Expression of Genes Involved in Saikosaponin Biosynthesis Between Bupleurum chinense DC and Bupleurum scorzonerifolium Willd. Front. Genet. 2020, 11, 583245. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, S.-R.; Xu, Y.-H.; Ge, Z.-Y.; Sui, C.; Wei, J.-H. Overexpression of BcbZIP134 negatively regulates the biosynthesis of saikosaponins. Plant Cell Tissue Organ Cult. (PCTOC) 2019, 137, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Jin, Y.; Huang, W.; Sun, Q.; Liu, F.; Huang, X. Full-length transcriptome sequences of ephemeral plant Arabidopsis pumila provides insight into gene expression dynamics during continuous salt stress. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.Z.; Jaroszewski, L.; Adam, G. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar]
- Amos, B.; Rolf, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar]
- Young, M.; Wakefield, M.; Smyth, K.G.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Chen, J.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar]
- Kong, L.; Yong, Z.; Ye, Z.Q.; Liu, X.Q.; Ge, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323–338. [Google Scholar]
- Anders, S. Analysing RNA-Seq data with the DESeq package. Mol. Biol. 2010, 43, 1–17. [Google Scholar]
- Xie, L.; Dong, C.; Shang, Q. Gene co-expression network analysis reveals pathways associated with graft healing by asymmetric profiling in tomato. BMC Plant Biol. 2019, 19, 373–384. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, J.L.; Thomas, D.S. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2002, 25, 402–408. [Google Scholar]
- Yu, M.; Liu, D.; Li, Y.C.; Sui, C.; Chen, G.D.; Tang, Z.K.; Yang, C.M.; Hou, D.B.; Wei, J.H. Validation of reference genes for expression analysis in three Bupleurum species. Biotechnol. Biotechnol. Equip. 2019, 33, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Stephen, G.H.; Richard, L. Terpenes, hormones and life: Isoprene rule revisited. J. Endocrinol. 2019, 242, 9–22. [Google Scholar]
- Fang, X.; Shi, L.; Ren, A.; Jiang, A.L.; Wu, F.L.; Zhao, M.W. The cloning, characterization and functional analysis of a gene encoding an acetyl-CoA acetyltransferase involved in triterpene biosynthesis in Ganoderma lucidum. Mycoscience 2013, 54, 100–105. [Google Scholar] [CrossRef]
- Lee, A.R.; Kwon, M.; Kang, M.K.; Kim, J.; Kim, S.U.; Ro, D.K. Increased sesqui- and triterpene production by co-expression of HMG-CoA reductase and biotin carboxyl carrier protein in tobacco (Nicotiana benthamiana). Metab. Eng. 2019, 52, 20–28. [Google Scholar] [CrossRef]
- Estevez, J.M.; Cantero, A.; Reindl, A.; Reichler, S.; Leon, P. 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J. Biol. Chem. 2001, 276, 22901–22909. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mutanda, I.; Wang, K.; Yang, L.; Wang, Y. Chloroplastic metabolic engineering coupled with isoprenoid pool enhancement for committed taxanes biosynthesis in Nicotiana benthamiana. Nat. Commun. 2019, 10, 4850–4861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ro, D.K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; He, Z.L.; Bi, X.C.; Xu, W.; Wei, T.; Wu, S.X.; Hu, S.N. Transcriptomic profiling reveals MEP pathway contributing to ginsenoside biosynthesis in Panax ginseng. BMC Genom. 2019, 20, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Wu, S.R.; Wang, L.; Xu, Y.H.; Wei, J.H.; Sui, C. Cloning and analysis of β-amyrin synthase gene in Bupleurum chinense. Genes Genom. 2015, 37, 767–774. [Google Scholar] [CrossRef]
- Zhao, C.; Xu, T.; Liang, Y.; Zhao, S.; Ren, L.; Wang, Q.; Dou, B. Functional analysis of β-amyrin synthase gene in ginsenoside biosynthesis by RNA interference. Plant Cell Rep. 2015, 34, 1307–1315. [Google Scholar] [CrossRef]
- Confalonieri, M.; Cammareri, M.; Biazzi, E.; Pecchia, P.; Fevereiro, M.P.S.; Balestrazzi, A.; Tava, A.; Conicella, C. Enhanced triterpene saponin biosynthesis and root nodulation in transgenic barrel medic (Medicago truncatula Gaertn.) expressing a novel β-amyrin synthase (AsOXA1) gene. Plant Biotechnol. J. 2009, 7, 172–182. [Google Scholar] [PubMed]
- Yu, J.K.; Zhang, D.B.; Yang, D.C. Biosynthesis and biotechnological production of ginsenosides. Biotechnol. Adv. Int. Rev. J. 2015, 33, 717–735. [Google Scholar]
- Augustin, J.M.; Kuzina, V.; Andersen, S.B.; Bak, S. ChemInform Abstract: Molecular Activities, Biosynthesis and Evolution of Triterpenoid Saponins. Phytochemistry 2011, 72, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Huang, S.W. Multi-omics data-driven investigations of metabolic diversity of plant triterpenoids. Plant J. 2019, 97, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Biazzi, E.; Carelli, M.; Tava, A.; Abbruscato, P.; Losini, I.; Avato, P.; Scotti, C.; Calderini, O. CYP72A67 Catalyzes a Key Oxidative Step in Medicago truncatula Hemolytic Saponin Biosynthesis. Mol. Plant 2015, 8, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S. Triterpene Structural Diversification by Plant Cytochrome P450 Enzymes. Front. Plant Sci. 2017, 8, 1886–1900. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, Y.; Wang, M. Discovery and Engineering of Cytochrome P450s for Terpenoid Biosynthesis. Trends Biotechnol. 2019, 37, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhao, S.J.; Wang, X.S. Functional regulation of a UDP-glucosyltransferase gene (Pq3-O-UGT1) by RNA interference and overexpression in Panax quinquefolius. Plant Cell Tissue Organ Cult. 2017, 129, 445–456. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ssa Content (µg/g) | Ssd Content (µg/g) |
|---|---|---|
| S1 | 0 | 0 |
| S2 | 0 | 0 |
| S3 | 444.56 | 312.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhao, J.; Chen, H.; Mao, Y.; Yang, Y.; Feng, L.; Mo, C.; Huang, L.; Hou, D.; Yu, M. Transcriptome Level Reveals the Triterpenoid Saponin Biosynthesis Pathway of Bupleurum falcatum L. Genes 2022, 13, 2237. https://doi.org/10.3390/genes13122237
Li Y, Zhao J, Chen H, Mao Y, Yang Y, Feng L, Mo C, Huang L, Hou D, Yu M. Transcriptome Level Reveals the Triterpenoid Saponin Biosynthesis Pathway of Bupleurum falcatum L. Genes. 2022; 13(12):2237. https://doi.org/10.3390/genes13122237
Chicago/Turabian StyleLi, Yuchan, Jun Zhao, Hua Chen, Yanping Mao, Yuping Yang, Liang Feng, Chuanxin Mo, Lin Huang, Dabin Hou, and Ma Yu. 2022. "Transcriptome Level Reveals the Triterpenoid Saponin Biosynthesis Pathway of Bupleurum falcatum L." Genes 13, no. 12: 2237. https://doi.org/10.3390/genes13122237