1. Introduction
White mold, caused by
Sclerotinia sclerotiorum (Lib.) de Bary, is considered one of the most economically important diseases affecting common bean (
Phaseolus vulgaris L.) in temperate growing environments [
1]. White mold is a soilborne, necrotrophic, and non-host-specific fungus that infects >400 plant species. In common bean, both the grain legume form (dry bean) and the vegetable form (snap or green bean) are strongly affected by this disease. In addition to reducing yield and quality in the field, snap bean harvest lots with >3% moldy pods may be rejected at the processing plant. Several cultural practices can be used to manage white mold in bean fields, such as decreased plant density, orientation of rows to prevailing winds, surface rather than overhead irrigation, the application of fungicides such as Fluazinam, and the use of biological controls such as
Coniothyrium minitans or
Trichoderma spp. [
2,
3]. However, the most effective strategy to reduce the disease severity and decrease the use of fungicides is breeding beans for host-plant resistance to white mold [
4].
The genetic control of resistance to white mold can be partitioned into physiological resistance and disease avoidance [
5]. Avoidance traits may allow for escape from the disease through early maturity before conditions are favorable for pathogen development or may create an unfavorable environment for the pathogen through altered growth habit and plant architecture. Disease avoidance includes traits such as canopy porosity, lodging resistance, reduced flower number, glabrous stems and leaves, and increased cuticle thickness [
5]. Plant architecture is controlled by both qualitative and quantitative factors and may be influenced by environmental conditions [
1,
5]. Physiological resistance to white mold in common bean appears to involve traditional pathogen defense pathways, in addition to oxalate tolerance [
6,
7]. Whereas some researchers have reported Mendelian inheritance, most have found quantitative resistance for physiological resistance [
4,
8,
9].
Many white mold resistance and avoidance quantitative trait loci (QTL) have been reported for common bean, most with a small to moderate effect and residing in all linkage groups except Pv10 [
1]. Twenty-seven QTL for disease resistance and 36 for avoidance have previously been identified in multiple biparental populations in independent studies using various testing methods [
5]. Previous research on 14 recombinant, inbred, biparental populations involving 12 dry bean and two snap bean parents identified 37 QTL associated with resistance. A meta-analysis of these populations revealed 17 distinct QTL [
9]. Recent genome wide association studies (GWAS) have refined the QTL to smaller intervals and identified new QTL [
10].
GWAS is a powerful genomics tool developed to detect the genes responsible for traits of interest in humans, animals, and plants [
11]. Several GWAS statistical models have been developed, including the general linear model (GLM), which considers fixed effects only, and the mixed linear model (MLM), which includes both fixed and random effects [
12]. Fixed and random circulating probability unification (FarmCPU) model appears to be particularly effective in controlling the false-positive error rate, which can be a problem with GLM, while reducing false negatives caused by excessive stringency of the MLM [
13]. In addition to FarmCPU, other last-generation GWAS models that improve type I and type II statistical errors include Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway (BLINK) and Settlement of MLM Under Progressively Exclusive Relationship (SUPER), which are generally considered to be comparable with FarmCPU [
14].
GWAS models must typically account for population structure to avoid a bias for false positives derived from population structure. Population structure in common bean is primarily determined by two regions of domestication: the Middle American center and the Andean center [
15]. Reproductive barriers exist between these two centers of domestication, which results in the expression of dwarf lethal alleles, seed unviability, and physiological abnormalities that are manifested when members belonging to one center of domestication are hybridized with members of the other center [
16]. Dry beans have largely maintained separation between the centers of domestication, but snap bean has undergone considerable hybridization, resulting in a high degree of admixture between these two distinct genetic populations [
17,
18]. Significant differences exist between the Middle American and Andean centers of domestication in terms of disease resistances and the genes that underlie domestication [
5,
19].
The objective of this research is to better understand the genetic basis of white mold resistance in snap beans by identifying QTL derived from (1) phenotypes from both a greenhouse setting and a field setting (2) using both a Middle American and Andean reference genome to assess novel QTL that may be present in only one reference genome. Snap beans have undergone selective pressures and extensive inter-gene pool introgression distinct from dry beans; the identification of these QTL will facilitate marker-assisted breeding in snap beans for white mold resistance, providing new sources of resistance for dry beans.
4. Discussion
The management of white mold disease through traditional breeding has always represented a significant challenge. Environmental conditions and their interaction with the genotype play an important role in disease development and progression, regardless of physiological resistance. It is generally understood that the straw test is an indicator of physiological resistance, whereas field tests measure physiological resistance in combination with disease avoidance [
5,
27,
28]. The relative importance of these two factors may vary from field trial to field trial depending on growing conditions and production methods. A comparison of the field and greenhouse data reveals significant differences. Differing SNPs are associated with disease severity in these two data sets, and the phenotypic data sets either do not correlate (in 2013) or show negative correlation (in 2012). Other studies of quantitative resistance to white mold have reported low or no correlation between field evaluations and the straw test [
5]. According to these differences, field conditions can involve a complex interplay of avoidance factors and physiological resistance. This interaction between genotype and environment may mask the QTL, with small a individual effect associated with physiological resistance.
We examined models with zero, one, two and generally found one PC axis to produce the best fit. A one- or two-PC-axis model makes biological sense and corresponds to the two centers of domestication. A separation of Middle American and Andean-derived accessions occurs along the first PC axis. A second PC axis represents a dispersion of Middle American accessions, varying from Oregon Blue Lake types at one extreme to European Extra Fine types at the other end of the axis, in addition to separating Andean snap beans that possess either T- or C-phaseolin seed storage protein. These may represent independent derivations of snap beans from dry beans, all within the respective centers of domestication [
18]. In this study, we found that two PC axes were over fitted and unnecessary.
In the DSI 2012 data set, three SNPs identified by GWAS had unusual allelic ratios. These SNPs were on Pv09 at 14,451,719 bp, Pv07 at 33,701,341 bp, and on Pv10 at 46,178,860 bp. The proportion of heterozygotes detected at these positions was 15% to 24% of the total. This was unexpected in a self-pollinating crop and may represent selective pressure for the heterozygous state or possibly an error in genotyping. None of these three SNPs has a large effect or considerable R2 value.
The physical map coordinates for some gene models shifted when the G19833 genome was updated to version 2.1, as reflected by the positions of the six copies of the gene models related to polygalacturonase-inhibiting protein 1 related gene models that exist in the common bean genome (
Table 9). This gene is important for white mold resistance [
29], although no copies were located near to associated SNPs detected in the present study. In this comparison of gene positions in three different references genomes, not only do the positions shift in all three, but the corrections from V1.0 of G19833 to V2.1 of G19833 can involve shifts between chromosomes (Pv09 to Pv11). Excluding this change of chromosome, the average shift in position for these six gene copies between version 1.0 and version 2.1 of G19833 is 650 kb. The differences in position between the G19833v2.1 genome and 5-593v1.0 range from 1 to 2 Mb. As such, the intervals in which the same gene model may reside may be closer to 500 kb–1 Mb rather than the 100 kb interval that has been used in common bean GWAS research.
The 5-593 and G19833 reference genomes represent different centers of domestication and therefore may have unique gene variants. The SnAP and SBDP panels contain lines from both centers, as well as many lines that are admixtures. Restricting the reference genome for GWAS to an Andean reference genome, such as G19833, may result in missing variants present in the Middle American reference genome, 5-593, and vice versa. In this study, we identified several unique SNPs present only in one of the two reference genomes, although matching identical QTL in both genomes represent a difficulty, owing to the differences in position and sequence. For example, although the SNPs found on Pv02 and Pv03 in both reference genomes for the DSI 2013 data set are likely the same QTL in both genomes (
Table 8), the alleles differ in terms of frequencies (A/C versus C/A for the Pv02 QTL) and are different alleles altogether (T/C vs. G/A for the Pv03 QTL).
A recent GWAS study of white mold resistance in a dry bean (
Phaseolus vulgaris) MAGIC population did not yield any overlaps with our study, although one QTL identified in the current study was adjacent to one reported in [
10]. This QTL is located on Pv04 at position 47,083,551 and is 979 kb from the SNP position found in the present study at 46,104,790. The absence of adjacent or overlapping QTL is not entirely surprising. The population used in the dry bean study had a narrow genetic base, consisting of the progeny from an intercross of eight elite pinto bean cultivars and advanced lines of Middle American origin, whereas we used a broad collection of commercial and heritage materials of both Andean and Middle American derivation. Moreover, the use of a relatively generous threshold of 0.1% to identify significant SNPs in the dry bean study may have resulted in the identification of SNPs that were filtered out in the present study by the more conservative Bonferroni cutoff.
A GWAS study of a diversity panel of Spanish common bean accessions produced 15 quantitative trait intervals (QTIs) located on six chromosomes [
30]. None of the QTIs in that study overlapped with our QTL, but several were adjacent, including QTI2_34, QTI3_0.9, QTI8_55, and QTI9_16 (
Table 10).
In addition to these GWAS studies and an unpublished M.S. thesis [
31], genetic mapping of white mold resistance in common bean has been conducted with recombinant inbred line (RIL) populations with relatively large QTL intervals. This work with RIL was summarized by Vasconcellos et al. [
9] using the G19833 genome v1.0. The shift between versions of the common bean genome is problematic in terms of making comparisons with the RIL work, but even with sizable shifts in position, it is reasonable to infer overlaps between the QTL identified in the RIL studies and those reported in the present study, owing to the considerably larger intervals of these QTL, with an average length of 5.2 Mb. Half of the straw test SNPs found in the current study overlap with the QTL found in previous RIL studies, and three significantly associated SNPs from the field data overlap (
Table 10).
A mix of SNPs ranging from low to relatively high SNP effects and R2 values were obtained in this study. This is to be expected for a quantitative trait with numerous contributing loci. This can be extrapolated further through genomic prediction utilizing all SNPs for model construction and selection, even if any individual SNP has an extremely small effect. This is a modeling approach that was not used in the present study, although some SNPs with very small effects were captured through GWAS. Future studies should examine the utility of genomic prediction compared to GWAS.
A high preponderance of PPR genes in close proximity to associated SNPs for white mold resistance has not been noted in previous studies. PPRs are not R genes but are recognized as disease resistance gene analogs [
32]. PPRs interact with chloroplasts and mitochondria through modification of organellar transcripts or their processing and translation [
33]. Chloroplasts are involved in immune signaling, and both chloroplasts and mitochondria are involved in the production of reactive oxygen species (ROS) and in programmed cell death [
32]. The loss of a mitochondrially targeted PPR in Arabidopsis considerably increased susceptibility to necrotrophic fungi from the loss of ROS homeostasis [
34]. Research into potato
Ralstonia infection revealed a differential upregulation of PPRs upon infection [
35]. Constitutive expression of a mitochondrially targeted PPR in a mutated rice plant enhanced disease resistance to both fungal and bacterial pathogens [
36]. Our research also revealed a high preponderance of LRR type genes, which have long been recognized as disease resistance gene analogs [
32,
37,
38]. LRRs are key molecules in both effector-triggered plant immunity and in pathogen/microbe-associated molecular pattern (PAMP)-triggered immunity [
32]. A quarter of the LRRs were STPKs, which are part of a class of transmembrane RLKs. These STPKs are some of the first molecules triggered by PAMP in an immune response cascade [
32,
39,
40]. Not all STPKs possess LRR motifs [
39], and two of those identified here did not but may still be involved in pathogen recognition. Collectively, nearly 80% of all SNPs identified in this study were near either a PPR, LRR, or RLK gene, suggesting that these types of defense genes may be of critical importance to building quantitative inheritance of white mold resistance in common bean, specifically snap bean.
The frequency of PPR genes identified in this study is not related to the frequency of this gene family in the genome. A keyword search of the G19833 genome, v2.1, reveals 514 copies of PPR, and a keyword search of the 5-593 genome, v1.1, yields 502 copies. At a total length of 537,218,636 bp, the G19833v2.1 genome would be expected to contain one PPR copy per 1.04 Mb. Likewise, the 5-593v1.0 genome is 572,228,222 bp in length, with one copy expected every 1.14 Mb. Our window for a candidate gene search was far smaller, at 200 kb, which should only yield 6 to 7 copies of the PPR gene if the average distribution were applied to our search windows; however, we found 15 copies near our associated SNPs.
Other defense-related genes were identified in this study. Two ankyrin genes were identified near associated SNPs. Ankyrin is known to be involved in PAMP immune recognition [
41]. A study in soybean revealed that
Fusarium infection upregulated ankyrin gene expression [
42]. A gain-of-function mutation in the SNC2 ankyrin gene resulted in constitutive defense responses in
Arabidopsis [
41]. Tandem chitinase genes were also found, and a modification of the chitinase gene or an enhancement of its transcription produced a more robust plant response in degrading chitin cell walls of the white mold fungus [
43,
44]. Ethylene-responsive transcription factors are also known to be important to plant defense. Ethylene signaling is critical for induced systemic resistance to necrotrophic fungal pathogens, and
Brassica napus infected with white mold showed induced ethylene pathway activity based on an analysis of transcriptome activity 48 h post infection [
7,
45]. In addition, zinc finger motif proteins, endoglucanase, subtilase, PMR5, peroxidase, thionin, xyloglucan, remorin, thaumatin, AT-hook, and lipid transfer proteins have all been implicated in plant defense [
32,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61].
There may be additional candidate genes not identified in this study. Some SNPs may be associated with avoidance traits, and we did not attempt to identify such candidates because avoidance constitutes is a very broad set of traits that are pleiotropically influenced by many genes. The SNP at 46,254,411 on Pv01 is near fin, which controls determinacy in common bean. Determinate growth habit has been associated with white mold avoidance, and we screened both determinate and indeterminate accessions in the greenhouse straw test in which this SNP was observed. However, we did not expect the greenhouse straw test to correlate with variation in avoidance traits. Finally, some SNPs might be associated with regulatory genes, and we did not search for these for similar reasons.

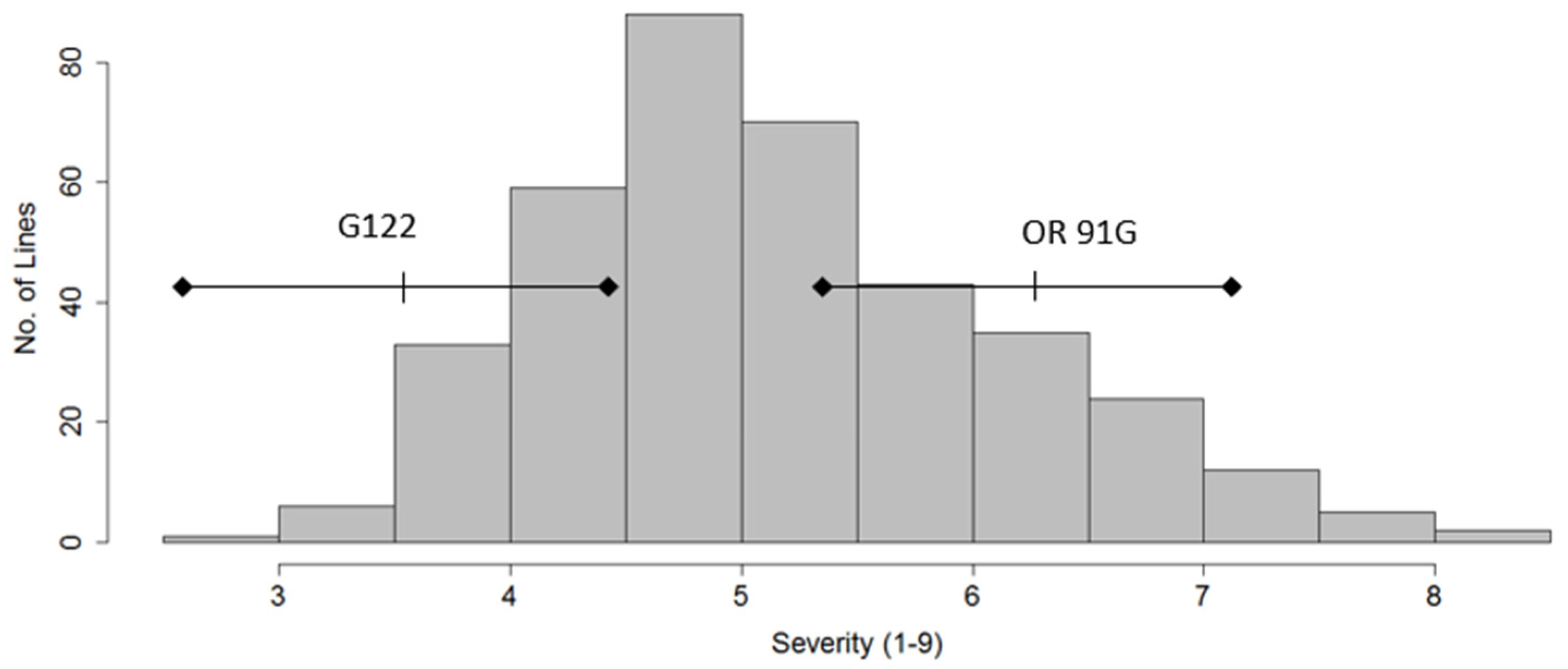
 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}