Abstract
The RNA methylation of adenosine at the N6-position (m6A) has attracted significant attention because of its abundance and dynamic nature. It accounts for more than 80% of all RNA modifications present in bacteria and eukaryotes and regulates crucial aspects of RNA biology and gene expression in numerous biological processes. The majority of m6A found in mammals is deposited by a multicomponent complex formed between methyltransferase-like (METTL) proteins METTL3 and METTL14. In the last few years, the list of m6A writers has grown, resulting in an expansion of our understanding of the importance of m6A and the methylation machinery. The characterization of the less familiar family member METTL16 has uncovered a new function of the m6A methylation apparatus, namely the fine-tuning of the cellular levels of the major methyl donor S-adenosylmethionine (SAM). METTL16 achieves this by adjusting the levels of the enzyme that synthesizes SAM in direct response to fluctuations in the SAM availability. This review summarizes recent progress made in understanding how METTL16 can sense and relay metabolic information and considers the wider implications. A brief survey highlights similarities and differences between METTL16 and the better-known METTL3/14 complex, followed by a discussion of the target specificity, modes of action and potential roles of METTL16.
1. m6A RNA Methylation
The regulation of gene expression through reversible chemical modifications of RNA, DNA and histone proteins is of paramount importance for normal development and differentiation. One of the most prominent modifications detected in mRNA is N6-methyladenosine [1,2]. Many recent studies have underscored that m6A methylation is also present in different types of non-coding RNAs [2,3,4,5,6] and plays important roles in diverse cellular processes, including stem cell differentiation and neurogenesis [2,7,8]. The dysregulation of m6A modification has been associated with aberrant animal development and several human diseases, notably cancer [8,9,10,11]. The functional consequences of m6A methylation are wide-ranging since m6A methylation has been linked to almost every step in RNA homeostasis. Effects on the RNA structure, RNA stability or effector protein binding by m6A in turn impact processes such as pre-mRNA splicing, mRNA export, translation initiation and chromatin activity, to name just a few [8,12,13,14,15,16].
Given the potential significance of RNA m6A modification in human health and disease, efforts of the past few years have focused on the identification of regulators of this modification. m6A methylation is introduced by methyltransferases and can be removed either passively, via the degradation of the modified RNA, or actively, by erasers such as the dioxygenases FTO or ALKBH5 [7,17,18,19]. Resembling the function ascribed to histone modifications, m6A modification can cause a structural switch and/or act as a signal for the recruitment of downstream effectors that influence the fate of the target mRNA [20,21,22,23,24]. Many functions of m6A are mediated through reader proteins. Some readers, such as YTH-domain-containing family members, can bind directly to m6A methylated RNAs. Alternatively, the modulation of the secondary RNA structure can expose or mask RNA-binding motifs recognized by reader proteins, leading to an m6A-dependent regulation of RNA maturation and gene expression [7,17,18,19].
2. m6A Writers
Active methyltransferases transfer a methyl group from the co-factor S-adenosylmethionine (SAM) to the substrate adenosine [25]. Table 1 illustrates that the five m6A methyltransferases identified to date are distinct and differ in several important ways. One, they generally target different RNAs and/or sites. For example, ZCCHC4 represents a 28S-RNA-specific methyltransferase [26] whereas METTL3/14 targets mRNAs, non-coding RNAs and primary micro-RNAs [5,20,27]. When a given RNA is modified by more than one writer, this usually involves different sites within the transcript due to distinct substrate and RNA recognition modes of m6A writers [25,28].

Table 1.
Specificity of m6A writers. Modified residue is underlined.
Two, different writers display different sequence motif preferences (Table 1), with some enzymes (METTL16; ZCCHC4) favoring a combination of sequence and structural features [28,31,35]. In contrast, METTL3/14 shows little dependency on a particular structure [29]. Three, whereas some RNA methyltransferases act in a complex (e.g., METTL3/14; METTL5) [25,32], others appear to function alone. In the METTL3/14 complex, METTL3 represents the catalytic subunit that binds SAM, whereas the catalytically inactive METTL14 promotes RNA-binding and stimulates methyltransferase activity [25,27,29,37,38]. Accessory factors such as Wilms tumor 1 associating protein WTAP or RNA-binding motif proteins RBM15/15B and others further modulate the METTL3/14 activity and specificity [17,38,39]. In contrast, METTL16 has been found to exist as a monomer or homodimer [28,40,41].
m6A methyltransferases harbor a signature motif of class I methyltransferases, the Rossmann fold (Figure 1a,b, MTFase domain), and, in most cases, additional domains that contribute to the regulation of the enzyme [25]. Zinc fingers in METTL3 constitute the RNA recognition domain and cooperate with the MTFase domains for catalysis (Figure 1a) [42]. Deletions of arginine/glycine motifs located in the C-terminus of METTL14 also reduce the RNA-binding affinity of METTL3/14 [43]. METTL16 recognizes its substrates via two domains that bear no resemblance to canonical RNA-binding motifs (Figure 1b). The unique N-terminus is required for RNA-binding and hence catalysis [28,31,41]. In higher eukaryotes, METTL16 additionally contains a C-terminal vertebrate-conserved region (VCR). An arginine-rich sequence within the VCR is critical for substrate binding and methylation. The precise function of this domain is still under investigation [31,44], but it has been shown that the VCR domain enhances the catalytic efficiency by lowering the Km by at least an order of magnitude [44]. In non-vertebrates, it is possible that accessory proteins take on the role of the VCR.
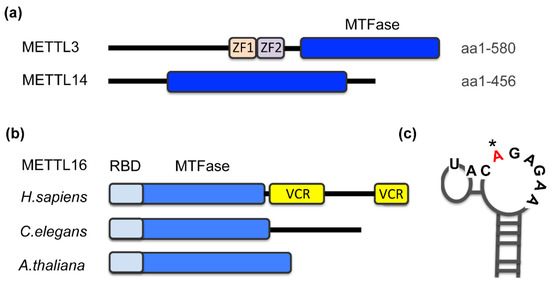
Figure 1.
Domain organization of m6A methyltransferases (a) METTL3, METTL14 and (b) METTL16. Schematic indicates domain architecture and function. MTFase, methyltransferase Rossmann fold; ZF, zinc finger domain; RBD, RNA-binding domain, VCR, vertebrate-conserved region. (b) METTL16 from selected species that are discussed in this review: human (aa 1-562), worm (METT-10) and Arabidopsis (FIONA1). (c) Stem-loop structure of a typical METTL16 target site with a specific nonamer sequence; the modified adenosine is highlighted (*). Based on the first hairpin of MAT2A 3′UTR.
The successful crystallization of METTL16, either of separate protein domains alone or in a complex with RNA, is extremely informative [28,31,41,44]. One outcome was the assignment of a regulatory role to a loop near the SAM-binding pocket that controls SAM-binding and hence methylation efficiency. Auto-inhibition via this so-called K-loop appears to be unique to METTL16 and is not observed in METTL3 [28]. Significant progress has also been made towards an understanding of the molecular basis of RNA- binding. Detailed accounts of these structural insights can be found here [25,45].
3. Multifaceted METTL16: Nuclear and Cytoplasmic, Catalytic and Non-Catalytic Roles
Much progress has been made in recent years in identifying RNAs bound by METTL16 [11,44,46,47,48,49,50,51,52,53]. It has emerged that METTL16 interacts with both coding and non-coding RNAs. These include, but are not limited to, small nuclear RNAs such as U6 [47,48], long non-coding RNAs [48,50] such as the cancer-related RNA RAB11B-AS1 [52] and the metastasis—associated lung adenocarcinoma transcript 1 (MALAT1) [46], as well as ribosomal RNA [48,50] and a set of mRNAs [48,53]. Representative examples belonging to different classes of RNAs, namely U6 snRNA, MALAT1 long non-coding RNA and MAT2A mRNA, have been validated as bona fide METTL16 RNA interactors [46,47,48].
One of the first identified and thus far best characterized METTL16 targets is the MAT2A mRNA [31,47,54], which encodes the key enzyme for methyl-donor synthesis in cells. It was subsequently shown that METTL16 is critical for preserving physiological SAM levels as discussed further below. In most cases, however, the function of the observed METTL16-RNA interaction is ambiguous as METTL16 can regulate the fate of its bound RNAs in diverse ways [45,55]. Like METTL3, the METTL16 protein acts in both the nuclear and cytoplasmic compartments, participating in RNA biogenesis, RNA decay and translational control [11,45,50,55]. Current models of how RNA methyltransferases might regulate different steps of gene expression include control through m6A modification. Accordingly, methyltransferase-activity-dependent functions of METTL16 have been documented, and have been shown to impact the mRNA stability and splice site choice [47,54,56]. However, one emerging concept is that not all METTL16-bound RNAs are methylated. This is based on the observation that, although METTL16 associates with thousands of RNAs, METTL16-catalyzed m6A methylation could be detected in only a small proportion of them [11,48]. The reasons could be, at least in part, technical, leading to an underestimation of methyl-sites. However, considerably more METTL16-bound than METTL16-modified RNAs were observed independent of the particular detection method applied or the type of RNA preparation used (e.g., total RNA versus nascent RNA) [11,48]. This favors a model in which METTL16, besides catalyzing the formation of m6A, exhibits significant methylation-independent functions [11,48]. In line with this interpretation, non-catalytic roles for METTL16 in both the nucleus and cytoplasm have been described [11,47,50,57]. For instance, METTL16 acts as a splicing enhancer of the mammalian MAT2A transcript independent of its methyltransferase activity [47]. Moreover, METTL16 prevents DNA-end resection in a methyltransferase-independent manner [57]. Cytosolic METTL16 promotes the efficient translation of thousands of transcripts independent of m6A through interactions with eukaryotic translation initiation factor 3a/b [11,50]. Potential contributions of METTL16 and m6A to cancer progression have been discussed elsewhere [9,10,11,57,58,59]. To sum up, it is becoming evident that METTL16 is a multifunctional enzyme with nuclear as well as cytoplasmic, catalytic and non-catalytic roles in gene regulation in physiological and pathological settings.
4. METTL16 Methylated RNAs
The extent to which METTL16 activity contributes to global m6A methylation remains an active area of research. Given that the entire field is young and flourishing, new technologies for high-confidence m6A mapping continue to be developed, allowing for the comparison of different datasets derived from m6A-antibody immunoprecipitation-based sequencing methods (e.g., m6A IP-seq or m6A-crosslinking exonuclease sequencing) and antibody-independent methods (e.g., metabolic labeling or small-molecule-based transcriptome editing) [11,47,49,51,53]. Collectively, these studies have pinpointed a few hundred direct m6A METTL16 candidate targets, including long ncRNAs, intronic sites and mRNAs associated with the DNA damage response, that await confirmation. A survey of these potential METTL16 methylation targets is provided by [55]. Currently, only two RNAs have been shown to be m6A METTL16 targets with any certainty: the mRNA MAT2A and the snRNA U6. Here, studies centered mainly around these two distinct transcripts are discussed to illustrate what is known about the substrate requirements and how METTL16 can regulate the fate of its target RNAs. METTL16 is, to date, the first and only RNA methyltransferase that acts as a metabolic sensor to safeguard SAM homeostasis. Therefore, emphasis will be on the functional significance of METTL16 in the maintenance of physiological SAM levels. Recent results from vertebrates and invertebrates will be reviewed that collectively reveal the principal ways that METTL16 regulates SAM biosynthesis through its catalytic and non-catalytic activities.
How might METTL16 selectively methylate certain transcripts and specific sites? It has been shown that the enzymatic activity of METTL16 is strictly dependent on a specific target sequence in combination with secondary structure features of the RNA (Table 1; Figure 1c). This conclusion is derived from a comprehensive in vitro and in vivo analysis of MAT2A and U6 RNA methylation sites [31,47,54,56] and holds true for independently characterized DNA-repair-related gene transcripts methylated by METTL16 [53]. Although one has to keep the inherent limitations of a very small sample size in mind, the results are exciting, revealing that METTL16 and METTL3/14 enzymes display very distinct substrate specificities. Whereas METTL3/14 exhibits activity towards single-stranded RNA with a “DRm6ACH” motif (in which D = A,G or U; R = A or G; H = A,G or U) [1,12,60], METTL16 preferentially methylates a nine-nucleotide consensus sequence UACm6AGARAA [28,31,47] (Table 1). However, this nonamer sequence only serves as an effective substrate when it is embedded in the appropriate secondary structure (Figure 1c). For methylation to occur, the target adenosine must be unpaired and flanked by stems, whereby nucleotides adjacent to the bulge influence the methylation efficiency significantly [28,31,56]. This information is frequently used to predict, based on sequence and structural context, which MTFase is responsible for a given m6A event in the transcriptome.
Of note, only a fraction of RNAs that contain the sequence consensus motifs for either METTL16 or METTL3/14 have been shown to be methylated [1,19,47]. It is possible that a subset of stimulus-dependent, dynamically regulated sites may have escaped identification [1,60]. Part of the explanation may also lie in the structural pre-requisite for METTL16 activity, since, for productive catalysis, the correct folding of the RNA may be necessary to properly present the target adenine residue to METTL16 [28]. In addition to the sequence and structure, extrinsic determinants are known to play an important role in shaping the m6A landscape controlled by METTL3/14 [12,17,61]. This is exemplified by trans-acting factors such as RNA-binding proteins or transcription factors that recruit METTL3/14 to promote the methylation of certain transcripts [12,19,58]. METTL16 is likely to similarly exploit protein co-factors to modulate its specificity and activity, possibly in a developmental or tissue-specific manner. To put this notion to test, several studies set out to purify METTL16 from mammalian cells and mouse tissues, aiming to identify candidate subunits and regulators [31,46,62,63]. Different experimental strategies were pursued to detect stable and transient interactions: either proximity-dependent labeling approaches or affinity purification after the precipitation of endogenous or tagged METTL16 coupled to mass spectrometry. Overall, remarkably few protein interactions were detected and, furthermore, these were largely mediated via RNA rather than direct protein–protein interactions [31,46,62,63]. Therefore, the model to date is that, in contrast to METTL3/14, METTL16 is not part of a stable protein complex and lacks other core subunits.
Protein–protein interaction networks often provide clues about the biological processes that the bait protein is engaged in. In the case of METTL16, there was little overlap between different datasets, but pre-mRNA splicing factors and U6 biogenesis factors were identified in a subset of the METTL16 interactomes [31,63]. The results of the follow up validation and functional experiments are eagerly awaited.
To what extent weak transient interactions or post-translational protein modifications (PTMs) are required for directing METTL16 function remain open questions. The phosphorylation of METTL16 induced by DNA damage was reported to result in decreased RNA-binding [57]. Other PTMs could fine-tune the subcellular localization of METTL16 or affect its stability, catalytic activity or protein interaction partners. What is clear is that the outcome of METTL16-deposited methylation can vary from the degradation of the mRNA (e.g., MAT2A) [47,54] to upregulation of gene expression (e.g., Brca2 mRNA) [53]. The relative contribution of canonical m6A reader proteins to the implementation of m6A signals set by METTL16 is an area yet to be explored.
Another conundrum is that, while only a small number of direct methylation targets of METTL16 have been confirmed, the depletion of this enzyme results in a genome-wide reduction in m6A [47,49]. This finding has important implications. It suggests that METTL16 has a broad impact on the m6A landscape through a combination of direct and secondary effects. Indeed, an in-depth motif analysis revealed that the vast majority of the affected sites are in fact METTL3–dependent [47,49]. This likely reflects the crucial role of METTL16 in SAM biosynthesis. Interference with the METTL16 function has been shown to cause SAM reduction [47]. In this way, METTL16 will directly impact methylation events catalyzed by METTL3, which depend on SAM availability [47,49,51]. Going forward, it will be essential to check the dependency of any given m6A site on both METTL16 and METTL3 to distinguish direct METTL16 targets. It will also be pertinent to determine the extent to which other RNA-, DNA- and protein methyltansferases that utilize SAM as a co-factor are affected by METTL16.
5. Timing of m6A Deposition and Its Position within the Transcript
Understanding how, when and where a modification occurs at a particular RNA residue is expected to provide clues to the functional significance of this modification. Efforts to determine the time point during the life cycle of a given RNA when m6A is installed by methyltransferases METTL3/14 and METTL16 have benefited from technological advances. One, m6A-antibody-based sequencing techniques have been applied not only to the total RNA but also to different cellular RNA fractions, comparing chromatin-associated, nuclear and cytoplasmic RNA pools. Such studies revealed that METTL3 deposits m6A co-transcriptionally on polymerase-II-transcribed pre-mRNA and chromosome-associated regulatory RNAs, primarily near terminal exons and within long internal exons of pre-mRNAs and at intergenic regions [64,65,66]. Similarly, METTL16 mainly installs m6A onto newly transcribed RNAs, including sites in the vicinity of the start codon, in exons and introns [11,48]. Two, an antibody-independent method (meCLICK-Seq) that relies on the catalytic activity of the enzyme under study likewise identified more m6A METTL16 sites in nascent than in mature RNAs [51]. This study revealed that over 75% of METTL16-dependent peaks fall within intronic regions, a much more significant proportion than for METTL3 [51] and than previously reported for METTL16 [11,48]. This difference to other METTL16 studies may reflect the fact that meCLICK-Seq was developed to map m6A in low-abundance transcripts derived from intronic and intergenic regions [51]. The authors further validated the intronic m6A marks in cell lines that carry deletions in selected intronic regions [51]. How might intronic m6A marks installed by METTL16 affect the fate of the transcript? One suggestion put forward is that they are related to intronic polyadenylation [51]. Given that intronic polyadenylation is widespread in cancer and usually leads to the generation of non-coding transcripts or truncated proteins [51], these findings provide a new and exciting direction for future investigations into the consequence of m6A marks set by METTL16.
6. METTL16 Methylates a Spliceosomal Component, but Does This Impact Splicing?
METTL16 binds and methylates U6 snRNA [31,47,48], a central component of the spliceosome transcribed by RNA polymerase III. Specifically, METTL16 deposits a single m6A methylation in a bulge in the stem of a hairpin structure in human U6 snRNA: the so-called ACAGA box [31,47,48]. This sequence lies in an evolutionarily conserved region important for splicing catalysis since it base-pairs with the 5′ splice site of pre-mRNAs in the first catalytic step of splicing [67,68]. In the past, investigations of the role of this highly conserved sequence motif in U6 snRNA have mainly focused on human and budding yeast, but with the discovery of METTL16 as the enzyme responsible for its methylation, researchers have recently began to look to other species. It was demonstrated that METTL16 orthologs in C. elegans and Arabidopsis represent the m6A writer for U6 snRNA, targeting an adenosine in the same sequence context as in human U6 (UACm6AGAGAA) [47,48,56,69,70]. In S. pombe, the METTL16 counterpart Mtl16 is responsible for the m6A modification at A37 in the ACAGA box; a mtl16 yeast deletion strain exhibits a loss of U6 snRNA methylation and slower growth rates [71]. Notably, in organisms with a small number of introns such as the budding yeast S. cerevisiae, U6 snRNA methylation is missing, and this correlates with the absence of METTL16 [47,71]. In conclusion, METTL16 represents the U6 snRNA methyltransferase that researchers were hunting for since the discovery of m6A in human U6 at position A46 over forty years ago [72,73].
What is the consequence of the METTL16 deposition of m6A into U6 snRNA? The frequency of this modification is nearly 100% and it occurs during early stages of U6 snRNP biogenesis [48]. U6 snRNA sits at the heart of the spliceosome, where it positions the substrate for the splicing reaction [74]. m6A could potentially impact the U6 snRNA function by modulating its stability or its interactions with RNAs and proteins. Methylated U6 snRNA gets incorporated into the U4/U6 snRNP, indicating that this methylation event is functional and possibly structural, and arguing against it being a target for a reader protein [48,74]. Mutations within the U6 snRNA ACAGA motif in yeast are lethal [75]. It is therefore tempting to speculate that the modification of this site impacts pre-mRNA splicing, but empirical evidence for this has proven surprisingly difficult to obtain. Global splicing defects are not readily evident in mammals or plants when METTL16 is depleted [31,53,70]. mettl16 null mutant mice embryos show little change in splicing patterns [31], but whether a maternal pool of methylated U6 snRNA can compensate for this is unresolved. Alternatively, other active RNA methyltransferases may be able to complement mettl16 mutants. However, this possibility seems less likely given that there is no evidence that different RNA methyltransferases can substitute for each other in vivo. A contribution of m6A U6 snRNA to the fine-tuning of the splice site selection is an attractive concept that requires more thorough investigation [31]. It is plausible that a loss of the U6 methyl-mark triggers subtle differences in the spliceosome assembly or affects the splicing of specific gene transcripts [48]. Indeed, a recent study demonstrates that the loss of U6 snRNA methylation in fission yeast [71] regulates the splicing of a subset of introns, especially those weakly recognized by U5 snRNA, another spliceosomal component [71]. This suggests that m6A in U6 can contribute to 5’ splice site recognition in a context-dependent manner. Based on these results, it will be exciting to revisit the question of whether the efficiency of the splicing of particular gene transcripts is affected by U6 snRNA m6A in metazoa.
While the biological significance of the METTL16 methylation of U6 snRNA is still debated, particularly for vertebrates, studies in mammals and worms have demonstrated that METTL16 post-transcriptionally regulates the expression of the key enzyme for the production of SAM. The mechanistic detail is different in vertebrates and invertebrates as surveyed below.
7. Roles of METTL16 in the Control of SAM Homeostasis
7.1. Introduction to SAM Synthetases (MATs)
The principal methyl-group donor of all cells, SAM, is generated from the amino-acid methionine and adenosine tri-phosphate (ATP) by methionine adenosytransferase (MAT) enzymes, referred to as SAM synthetases (Figure 2) [76,77]. SAM is required for transmethylation reactions of RNA, DNA and proteins, as illustrated in Figure 2, but also for polyamine and glutathione biosynthesis [76,78,79,80]. Because of the pivotal position of methionine in the metabolic network of the cell, the consequences of methionine availability for cells and organisms are far-reaching, with many mechanisms and signaling pathways feeding into and contributing to the tight control of physiological SAM levels [78,80]. The purpose of this review is to highlight one particular aspect of SAM regulation, which controls gene expression in response to metabolic fluctuations through the regulation of RNA metabolism.
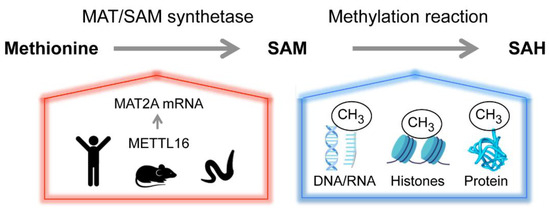
Figure 2.
Homeostasis of the versatile methyl donor S-adenosyl-methionine (SAM or AdoMet). Methionine adenosyltransferase (MAT or SAM synthetase) catalyzes the formation of SAM from methionine and ATP. Methyltransferases transfer the methyl group from SAM to a variety of acceptor molecules (e.g., nucleic acids, proteins and lipids). The product, S-adenosylhomocysteine (SAH), is recycled to regenerate methionine but can also act as an allosteric regulator. SAM contributes to several biosynthetic pathways and is central to many cellular functions, including epigenetic regulation, cell growth and maintaining the redox status of the cell. The physiological levels of SAM are tightly controlled. In human, mouse and C. elegans, the m6A RNA methyltransferase METTL16 senses SAM and controls the abundance of SAM synthetase MAT2A mRNA post-transcriptionally.
A strategy commonly used by cells to sense and respond to actual methionine levels is to monitor SAM levels [80]. SAM synthetases, which are highly conserved from prokaryotes to humans, lie at the heart of this regulation [81,82]. Mammalian systems have two distinct catalytic subunits, MAT1A and MAT2A [78,83,84]. MAT1A is mainly found in hepatocytes, whereas MAT2A is ubiquitously expressed and is complexed with a regulatory subunit, MAT2B [78]. Early studies found that the amount of MAT2A is inversely correlated with the cellular methionine concentration, where a decrease in methionine causes an increase in the amount of MAT2A [85,86,87]. Transcriptional and post-transcriptional mechanisms were reported to effect MAT2A levels in the liver [78,81,83,87,88]. Interestingly, both eukaryotes and prokaryotes exploit RNA regulatory elements located within the MAT transcripts to control their expression in response to the need for SAM. RNA structures in SAM synthetases of prokaryotes and fission yeast bind the metabolite directly [89,90]. Bacterial riboswitches represent the paradigm for this concept [89,90]. Here, the direct binding of SAM induces a structural switch in the RNA, which results in the inhibition of transcription and/or translation [89,91,92]. In contrast, in higher eukaryotes, SAM indirectly regulates mRNA levels of SAM synthetase genes. For example, the RNA-binding protein HuR is involved in MAT2A RNA stability control in liver cells [81]. Mechanistically, methylated HuR causes MAT2A mRNA decay [81]. Our understanding of how the regulation of MAT synthetase transcripts can be achieved in other cell types received a boost when recent studies brought a new player into focus: the m6A methyltransferase METTL16 [47,54]. It provides an elegant mechanistic explanation of how nutrients and metabolic conditions, previously acknowledged to influence the epigenetic status of a cell, can impact gene regulation via the epitranscriptome in a highly integrated process.
7.2. METTL16 in Mammals Governs SAM Synthetase
Six METTL16 consensus methylation sites are present in the 3′ untranslated region (UTR) of MAT2A mRNA, each one positioned in a stem loop and validated as a METTL16 substrate (Figure 3c, left) [47,48,49,93]. These hairpin structures are vital for ensuring the optimal production of SAM synthetase in response to changing SAM levels. The recruitment of METTL16 to these hairpins triggers two distinct events. The first entails the modulation of the MAT2A pre-mRNA splicing pattern in an m6A-independent way. The other m6A-dependent mechanism involves the regulation of MAT2A mRNA stability. Remarkably, SAM levels determine the precise METTL16 function as described in detail below.
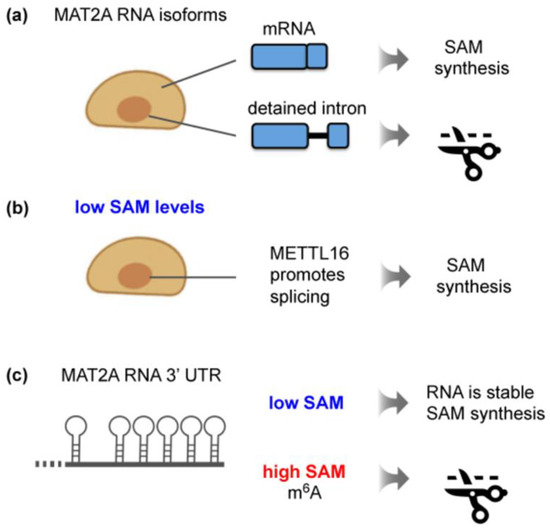
Figure 3.
Regulation of mammalian MAT2A expression by METTL16 in response to intracellular SAM levels. (a) Mammalian MAT2A has two transcript isoforms: a cytoplasmic mRNA and a nuclear, unproductive isoform with a detained last intron. (b) Stable METTL16 binding to the 3′UTR of MAT2A induces full splicing of MAT2A pre-mRNA, leading to increased SAM synthesis. This effect is independent of m6A deposition by METTL16. (c) The 3′UTR of MAT2A contains six hairpins. Their m6A methylation by METTL16 results in SAM-responsive RNA degradation of MAT2A mRNA. Given that the 3′UTR hairpins of MAT2A mRNA are found exclusively in vertebrates [93], a different mechanism must operate in lower eukaryotes.
Extensive studies of MAT2A in mammalian cells have reported the existence of a nuclear transcript isoform that is incompletely spliced [47,86]. This detained-intron MAT2A transcript is subject to nuclear degradation (Figure 3a). A series of elegant experiments from the group of N. Conrad uncovered that, when the SAM supply becomes limiting, METTL16 binding to the 3′UTR enhances the efficiency of co-transcriptional splicing (Figure 3b). This shifts the balance towards the production of mature MAT2A mRNA, ultimately increasing the production of MAT2A protein [47,94]. Intriguingly, it was demonstrated that METTL16 enzymatic activity itself is not required for the induction of MAT2A splicing [47]. In fact, catalytically dead METTL16 promotes the splicing of a reporter MAT2A construct irrespective of the SAM concentration [28]. Here, a single amino acid exchange (N184A) in the SAM-binding site abrogates the methyltransferase activity but retains the RNA-binding capacity of METTL16 [28]. A hyperactive enzyme (K163A), on the other hand, does not enhance splicing [28]. The critical parameter is arguably the dwell time of METTL16 on the 3′UTR, although direct biophysical measurements have not been provided. Low SAM levels were proposed to lead to a decreased enzymatic turnover of METTL16, resulting in an increased residence time [47]. The prolonged binding of METTL16, especially to the hairpin proximal to the intron, stimulates the efficient splicing of MAT2A transcripts. Consistent with this notion, a screen for factors required to induce the splicing of a synthetic MAT2A reporter construct upon methionine depletion identified METTL16. Besides METTL16, other hits were the co-activator of RNA polymerase II MED9 and the cleavage factor I subunit CFIm25 (NUTDT21) [95]. From recent studies, CFIm25 has emerged as a regulator of several RNA-processing events other than polyadenylation through preferential binding to UGU-containing sequences in RNAs [96]. Tethering assays suggest that the association of the CFIm complex with the detained intron and the 3′UTR drives MAT2A splicing [95]. This occurs downstream of METTL16 binding, but how these two events are linked is not understood. Notably, splicing regulation relies on an intact VCR domain of METTL16 [47]. Given that this domain associates with RNA, it was speculated that prolonged METTL16 binding may trigger an RNA conformational change that exposes binding sites for splicing factors [44,47].
Although there is no documented role for m6A in the stimulation of MAT2A splicing in mammals, the methylation function of METTL16 is crucial for controlling the steady-state levels of mature MAT2A mRNA (Figure 3c) [28,47,54]. The following findings reveal m6A methylation as an integral step in SAM regulation in mammals. When SAM is abundant, an increased METTL16-induced methylation of the hairpins located in the 3′UTR occurs, triggering the destabilization and degradation of the transcript [47,54]. Conversely, an increase in MAT2A mRNA levels observed upon methionine depletion correlates with low m6A levels in the 3′UTR of MAT2A [47,54,85]. Consistent with this mechanism of action, catalytically inactive METTL16 causes MAT2A mRNA stabilization, whereas hyperactive METTL16 results in reduced MAT2A mRNA levels [28]. It is not yet clear how METTL16-methylated MAT2A is recognized for degradation and whether known m6A reader proteins are involved. Based on experiments with reporter constructs, components of the general m6A machinery, such as the reader YTHDC1 and the demethylase FTO, have been implicated in MAT2A mRNA stability control, but understanding the mechanisms requires further exploration [54]. In general, the methylation of MAT2A by METTL16 provides a notable example of an RNA modification that is established in response to a metabolic cue and, in turn, regulates the expression of the target transcript.
Since mammalian METTL16 can directly influence the methyl-donor capacity of the cell, one would anticipate that the depletion of METTL16 impacts the methylome and transcriptome. Accordingly, it was reported that the Mettl16 gene is essential for the survival of the vast majority of human cancer cells, for mouse development and in adult mice [11,31,56]. It has proved impossible to bring viable Mettl16 or Mat2a null mice to term [31,56,97]. Knock-in mouse mutants further revealed that both the catalytic activity and the RNA-binding capacity of METTL16 are essential for development [31,56]. The time of death of Mettl16 knockout embryos is around implantation [31]. Strikingly, at embryonic day 2.5 (E2.5) at the morula stage, embryos that lack Mettl16 show little alteration in their gene expression, one exception being the Mat2a transcript, which is significantly downregulated. This picture changes dramatically in E3.5 blastocysts, which show massive transcriptome dysregulation accompanied by developmental arrest. These data lead to the conclusion that METTL16 activity is essential for embryonic development through the regulation of Mat2a mRNA levels and thus SAM availability [31].
At this stage, our understanding of the consequence of METTL16 deficiency on the epigenome is limited. One can only speculate that the main cause of the death of Mettl16 null mice could be either the dysregulation of a single, major event or the sum of several disturbances of metabolic and epigenetic pathways crucial for normal development. Due to the complexity of the processes involved, probing and interpreting the causalities is challenging, but chromatin dynamics may represent a suitable starting point for further interrogation. DNA methylation is undoubtedly the best-understood epigenetic modification in the early development and inactivation of major components of the DNA methylation machinery results in embryonic lethality [98]. At the blastocyst stage, the bulk of genomic DNA is hypomethylated, with the exception of imprinted genes and retrotransposons, whose methylation is maintained [98]. As the embryo implants in the uterus, DNA methylation is widely re-instated [98]. It is possible that METTL16 is involved in this extensive DNA methylation reprogramming. Therefore, it will be exciting to directly investigate whether DNA methylation is one of the processes that become disrupted in the Mettl16 mutants and whether it is maintenance and/or de-novo methylation, which is deficient. Histone methylation may also be affected, although pilot experiments in human cells lacking METTL16 have not provided supporting evidence [11]. A future systematic genome-wide interrogation of the potential impact on histone and DNA methylation in human and mouse cells lacking METTL16 should provide answers to the question of whether fluctuations in SAM levels in response to a loss of METTL16 are sufficient to alter the epigenetic landscape.
Collectively, these studies in mammals postulate that METTL16 can act as an SAM sensor, but the generality of this is not clear. It is therefore important to turn to other model organisms such as invertebrates and further interrogate the relevance of the catalytic activity of METTL16.
7.3. METTL16 in Nematodes Regulates SAM Synthetase Pre-mRNA Splicing via m6A
It turns out that SAM production in nematodes is also modulated by METTL16 [56,99]. This involves m6A methylation catalyzed by METTL16, which, in turn, affects pre-mRNA splicing. It is worth stressing that METTL16 impacts MAT2A splicing in different ways in mammals and in worms. One, the splice events occur at different locations in the MAT2A transcript. Two, the splicing of mammalian MAT2A pre-mRNA involves METTL16 binding but not methylation. Therefore, m6A has no apparent role in this particular splicing phenotype in mammals. In contrast, m6A is central to the MAT2A-splicing phenotype in worms. The mechanistic details were uncovered by taking advantage of the fact that nematodes lack the METTL3/14 m6A writer complex dominant in mammals [56,100], making them an ideal model to study the molecular and physiological effect of METTL16. The C. elegans mettl16 ortholog, mett-10, is required for normal development [40]. Although the METT-10 enzyme lacks the vertebrate specific C-terminus it methylates similar substrates as its mammalian counterpart, particularly U6 snRNA and sams-3, sams-4 and sams-5 transcripts, the C. elegans orthologs of mammalian MAT2A [56,99]. Notably, the number and location of the methylation sites in the SAM synthetase transcripts differ between mammals and worms [56]. This is not surprising since sams transcripts do not contain the 3′UTR hairpins conserved in MAT2A mRNAs of vertebrates [93]. Instead, a single m6A is present in worm SAM transcripts in a nonamer sequence that closely resembles the mammalian consensus, UACm6AGAaAc (lower case indicates worm specific bases), and is predicted to fold into a stem-loop structure. Intriguingly, the modified adenine base within this motif is at a location known to play a major role in splice site selection: at the invariant AG dinucleotide at the 3′ end of an intron. Two landmark studies uncovered that the methylation of this adenosine residue inhibits the use of this particular splice site, ultimately controlling the steady-state level of SAM synthetase [56,99]. How is this achieved given that the C. elegans genome does not code for orthologs of the YTH family of m6A readers or demethylases [100]? It was shown that the methyl-mark set by METT-10 precludes the binding of the essential splicing factor U2AF35, preventing spliceosome assembly [56,99,101]. As a consequence, alternative splicing coupled with nonsense-mediated mRNA decay occurs, leading to reduced levels of the SAM synthetase [56,99]. This model was developed to explain observations of m6A immunoprecipitates being enriched in the non-productive sams isoforms but depleted for the correctly spliced, productive mRNA [99]. On the other hand, an increase in the correctly spliced, productive sams isoform was observed in mett-10 mutant worms [56,99]. Using transgenic worms, it was further confirmed that exonic mutations that abolish 3′ slice site m6A methylation in vitro allow for efficient splicing in vivo [56].
A link between m6A and pre-mRNA splicing has been appreciated for years in many model organisms, especially in Drosophila [19]. One challenge in mammals has been to distinguish between direct versus indirect roles of m6A in splicing control. The described studies in worms provide the first demonstration that the presence of an m6A modification at a 3′splice site can directly interfere with splicing. Splice sites and the mechanisms of their recognition are highly conserved across the animal kingdom. This prompted Mendel et al. [56] to investigate whether the human splicing machinery is similarly sensitive to the presence of an m6A. In a series of elegant experiments, they artificially introduced an m6A either at a 3′ splice site or into an unrelated exon sequence of a human reporter construct and performed splicing assays in HeLa cell extracts. Exonic methylation did not inhibit splicing whereas methylation of the 3′ splice site did. Using a computational approach, putative 3′ splice site targets for mammalian METTL16 were identified. While mouse METTL16 was found to have the potential to methylate these 3′ splice sites in vitro, data on the in vivo significance are missing [56]. The jury is thus still out as to whether alternative splicing control by means of m6A methylation of an 3′splice site is the conserved principal mechanism beyond invertebrates. Interestingly, intronic polyadenylation has recently been linked to METTL16 methylation activity in mammals [51], suggesting that m6A deposited by METTL16 can determine the choice of alternative pre-mRNA-processing events in various ways.
Crucially, methylation and the alternative splicing of nematode SAM synthetase transcripts are linked to nutrient levels [56,99], highlighting the capacity of METT-10 to sense and respond to nutrient availability. In response to high SAM levels in nutrient-rich media, METT-10 installs m6A at a splice site to inhibit productive splicing and hence SAM synthetase production. Under low-nutrient conditions, m6A is absent, allowing for efficient splicing and the production of functional SAM synthetase, which, in turn, can generate more SAM from methionine and ATP. Accordingly, SAM synthetase activity autoregulates the expression of SAM synthetase genes in worms in response to nutrient availability through alternative splicing involving METT-10 enzyme activity. To understand the functional implications, mutants that interfere with the mett-10 function and SAM synthesis were analyzed and shown to have fertility defects [56]. What is missing is information on whether epigenetic pathways are disrupted in mett-10 mutant worms. C. elegans lacks DNA methylation but it has been shown that the depletion of sams-3 and sams-4 globally reduces histone methylation and disrupts heterochromatin organization [102]. This demonstrates that normal SAM levels are critical for maintaining the C. elegans epigenome and implies that m6A methylation deposited by METTL16 could play a significant part in this regulation.
How conserved is this mechanism of controlling cellular SAM levels via alternative splicing of the SAM synthetase pre-mRNAs in other invertebrates? At the center of this regulation lies a nonamer sequence that is recognized by METT-10 when imbedded within a stem loop structure at an exon-intron border and methylated in response to a rich diet [56,99]. Based on the fact that this sequence is conserved among worms, silk moth and flies and can indeed be methylated by METTL16 in vitro [56] it has been proposed that this type of SAM synthetase regulation may be widely used among invertebrates.
7.4. METTL16 in Plants and Fission Yeast Has Not Been Implicated in SAM Homeostasis
Because MAT2A is an established target of METTL16 in the vertebrates and invertebrates studied thus far, the question pertains as to whether other species use METTL16 to regulate SAM synthetase expression. METTL16 is highly conserved across many metazoans, as well as in plants, fission yeast and bacteria [31,44,47,55]. Of these representative species, METTL16 has been functionally characterized in mammals, C. elegans, Arabidopsis thaliana and fission yeast. S. pombe possesses a single gene for SAM synthetase, sam1. The fission yeast METTL16, Mtl16, neither methylates SAM synthetase RNA nor does its deletion affect the transcript levels of sam1 [71]. The sam1 transcript harbors a tertiary structure in its 5′UTR, which, upon binding directly to the SAM molecule, mediates repression of translation [90]. This is analogous to the ligand-sensing sam1 mRNA in bacteria, which regulates the SAM metabolism in a negative feedback cycle [89].
The Arabidopsis METTL16 ortholog is called FIONA1 (encoded by At2g21070). Originally described as a regulator of circadian rhythms [103], FIONA1 was very recently shown to be a bona fide RNA methyltransferase [69,70]. Plants, unlike worms, additionally contain the m6A methyltransferase Mettl3/14 enzyme complex, termed MTA (encoded by At4g10760) and MTB (encoded by At4g09980), that is responsible for the vast majority of m6A methylation [104,105]. Knockout mutations in the MTA/MTB genes are embryonic lethal in Arabidopsis [59,60], underscoring the importance for m6A RNA methylation in plant development. The disruption of FIONA1, on the other hand, results in early flowering and, at the molecular level, in a mild decrease (10–15%) in global m6A levels, which, in turn, can be restored by the expression of active FIONA1 [69,70]. The U6 splicing snRNA, as well as a small subset of mRNAs, have been identified as FIONA1-specific m6A target sites [69,70]. The functional consequences of FIONA1-dependent methylation involve the regulation of transcript abundance and alternative polyadenylation [69,70].
SAM deficiency suppresses the methylation of DNA and histones in rice, leading to a late-flowering phenotype [106]. However, how SAM synthetase activity is controlled is not understood as the regulatory subunit MAT2B is lacking in plants [107]. The MAT2A catalytic subunit is encoded by the MAT1-4 genes in Arabidopsis [107]. The corresponding transcripts have detectable m6A methylation but whether it is installed by FIONA1 is controversial; two groups disagree on whether methyl marks in MAT1-4 are reduced when FIONA1 function is disrupted [69,70]. The reasons for this discrepancy could be technical, given that seedlings of different ages were investigated by different methods: either m6A- or nanopore sequencing. Nevertheless, the consensus from both studies is that the expression levels of MAT1-4 transcripts are not affected by FIONA1, neither under normal nor high-SAM conditions [69,70]. Whether FIONA1 regulates MAT transcript processing in different ways—for instance, at the level of localization or translation—has not been investigated. FIONA1 is not essential for viability [69], which may argue against this enzyme being a key modulator of SAM levels. On balance, the evidence to date does not provide strong support for the role of FIONA1 in the regulation of SAM homeostasis. It remains to be seen whether other RNA methyltransferases contribute to the control of SAM synthetase activity in plants or what alternative mechanisms exist.
8. Concluding Remarks
METTL16 is a versatile RNA-binding and modifying enzyme engaged in the control of splice site selection, RNA stability and translation [11,31,47,54,56,58,71,95]. Through its multlifunctionality it influences various cellular processes, including the maintenance of genome integrity, proliferation, erythropoiesis and cancer progression [11,52,53,57,59,80,108]. Future efforts to understand these diverse roles will benefit from the identification of METTL16 regulators and effectors that, so far, have remained largely elusive. One of the best-understood and fundamental functions of METTL16 is the fine-tuning of MAT2A expression to regulate cellular SAM levels in humans, mice and nematodes [28,31,47,56,94,99]. To achieve this, METTL16 drives self-sustaining feedback loops that link MAT2A transcript abundance with SAM synthesis. It will now be important to investigate METTL16 function in MAT2A regulation in pluripotent stem cells and cancer models, as these cell types have an unusually high dependence on methionine, and altered metabolism is a hallmark of cancer [79,109,110,111,112,113]. Work in human and animal models has documented that changes in SAM levels can impact chromatin organization and actively contribute to the regulation of transcriptional programs [109,110,114,115,116,117]. To what extent the METTL16-dependent regulation of SAM synthesis correlates with epigenetic changes in DNA and histone methylation and under which circumstances it impacts chromatin states remains to be determined.
The physiological strategy to utilize RNAs for the regulation of SAM homeostasis is interesting in light of the fact that RNA transcripts turn over and RNA modifications will be lost upon turnover. This may allow for a swift yet relatively transient and therefore flexible response to changing environmental conditions. The involvement of multiple METTL16 target stem loops in MAT2A regulation in mammals is particularly intriguing as it suggests a co-operative mechanism for sensing SAM levels [54] rather than a simple on and off switch. One could envision a model in which the number of sites methylated by METTL16 at any one time allows for fine control, like a dimmer switch. Structural studies are in line with this idea, indicating that different hairpins are methylated with varying efficiencies when using pure enzymes [28]. Such a cooperative mechanism would provide a sophisticated regulatory response to fluctuating SAM levels.
Overall, the results reviewed here draw attention to METTL16 as a paradigm of an RNA methyltransferase at the intersection of metabolism and gene regulation. One principle that has emerged is that, in mammals and worms, METTL16/METT-10-mediated methylation events turn SAM production down in response to high intracellular SAM levels. In mammals, the regulation is more sophisticated, with METTL16 additionally ensuring an increase in SAM production when the demand rises. The mechanistic detail is revealing because, in the absence of SAM, METTL16 binds stably to unmodified MAT2A RNA and performs a non-catalytic function. Therefore, METTL16 can be considered as a writer and a reader. These distinct METTL16-dependent layers of SAM regulation observed in mammals are reminiscent of belt and braces and likely part of an intricate, multi-layered regulatory network preserving physiological SAM levels. An added complexity arises from the fact that SAM is interconnected with other metabolic pathways that affect nearly all aspects of cellular physiology [78,80], making it challenging to demonstrate causality. At the same time, this opens many avenues for further exploration and will likely drive the development of new concepts that will advance our understanding of the interplay between chromatin, RNA and metabolic networks in genome regulation.
Funding
This research received funding from the DFG TRR81/3-2022 project number 109546710.
Informed Consent Statement
Not applicable.
Acknowledgments
I would like to thank my colleagues for discussion and comments.
Conflicts of Interest
The author declares no conflict of interest.
References
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Sergiev, P.V.; Aleksashin, N.A.; Chugunova, A.A.; Polikanov, Y.S.; Dontsova, O.A. Structural and evolutionary insights into ribosomal RNA methylation. Nat. Chem. Biol. 2018, 14, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Piekna-Przybylska, D.; Decatur, W.A.; Fournier, M.J. The 3D rRNA modification maps database: With interactive tools for ribosome analysis. Nucleic Acids Res. 2008, 36, D178–D183. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- An, Y.; Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 2022, 21, 14. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; He, P.C.; Liu, W.; Wei, J.; Zhao, Z.; Gao, L.; Han, L.; et al. METTL16 exerts an m6A-independent function to facilitate translation and tumorigenesis. Nat. Cell Biol. 2022, 24, 205–216. [Google Scholar] [CrossRef] [PubMed]
- He, P.C.; He, C. m6 A RNA methylation: From mechanisms to therapeutic potential. EMBO J. 2021, 40, e105977. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Chen, J. The Biogenesis and Precise Control of RNA m6A Methylation. Trends Genet. 2020, 36, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Fang, Z.; Mei, W.; Qu, C.; Lu, J.; Shang, L.; Cao, F.; Li, F. Role of m6A writers, erasers and readers in cancer. Exp. Hematol. Oncol. 2022, 11, 45. [Google Scholar] [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Spitale, R.C.; Flynn, R.A.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.-W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucl. Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N(6)-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J. Mol. Biol. 2016, 428, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Oerum, S.; Meynier, V.; Catala, M.; Tisné, C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021, 49, 7239–7255. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Cai, J.; Dai, Q.; Natchiar, S.K.; Lv, R.; Chen, K.; Lu, Z.; Chen, H.; Shi, Y.G.; et al. N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2019, 15, 88–94. [Google Scholar] [CrossRef]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef]
- Doxtader, K.A.; Wang, P.; Scarborough, A.M.; Seo, D.; Conrad, N.K.; Nam, Y. Structural Basis for Regulation of METTL16, an S-Adenosylmethionine Homeostasis Factor. Mol. Cell 2018, 71, 1001–1011.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Mendel, M.; Chen, K.-M.; Homolka, D.; Gos, P.; Pandey, R.R.; McCarthy, A.A.; Pillai, R.S. Methylation of Structured RNA by the m6A Writer METTL16 Is Essential for Mouse Embryonic Development. Mol. Cell 2018, 71, 986–1000.e11. [Google Scholar] [CrossRef] [PubMed]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucl. Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, V.V.; Stolz, P.; Kaiser, S.; Gustafsson, T.H.; Lastres, P.R.; Sanz-Moreno, A.; Cho, Y.-L.; Amarie, O.V.; Aguilar-Pimentel, A.; Klein-Rodewald, T.; et al. The rRNA m6A methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 2020, 34, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Rong, B.; Zhang, Q.; Wan, J.; Xing, S.; Dai, R.; Li, Y.; Cai, J.; Xie, J.; Song, Y.; Chen, J.; et al. Ribosome 18S m6A Methyltransferase METTL5 Promotes Translation Initiation and Breast Cancer Cell Growth. Cell Rep. 2020, 33, 108544. [Google Scholar] [CrossRef]
- Ren, W.; Lu, J.; Huang, M.; Gao, L.; Li, D.; Wang, G.G.; Song, J. Structure and regulation of ZCCHC4 in m6A-methylation of 28S rRNA. Nat. Commun. 2019, 10, 5042. [Google Scholar] [CrossRef]
- Pinto, R.; Vågbø, C.B.; Jakobsson, M.E.; Kim, Y.; Baltissen, M.P.; O’Donohue, M.-F.; Guzmán, U.H.; Małecki, J.M.; Wu, J.; Kirpekar, F.; et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 2020, 48, 830–846. [Google Scholar] [CrossRef]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef]
- Dorsett, M.; Westlund, B.; Schedl, T. METT-10, a putative methyltransferase, inhibits germ cell proliferative fate in Caenorhabditis elegans. Genetics 2009, 183, 233–247. [Google Scholar] [CrossRef]
- Ruszkowska, A.; Ruszkowski, M.; Dauter, Z.; Brown, J.A. Structural insights into the RNA methyltransferase domain of METTL16. Sci. Rep. 2018, 8, 5311. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dong, X.; Gong, Z.; Qin, L.-Y.; Yang, S.; Zhu, Y.-L.; Wang, X.; Zhang, D.; Zou, T.; Yin, P.; et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N6-methyladenosine methyltransferase. Protein Cell 2018, 10, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Schöller, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA 2018, 24, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Yamashita, S.; Tomita, K. Mechanistic insights into m6A modification of U6 snRNA by human METTL16. Nucl. Acids Res. 2020, 48, 5157–5168. [Google Scholar] [CrossRef]
- Ruszkowska, A. METTL16, Methyltransferase-Like Protein 16: Current Insights into Structure and Function. Int. J. Mol. Sci. 2021, 22, 2176. [Google Scholar] [CrossRef]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 14013–14018. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e14. [Google Scholar] [CrossRef]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Höbartner, C.; Sloan, K.E.; Bohnsack, M.T. Human METTL16 is a N6-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef]
- Koh, C.W.Q.; Goh, Y.T.; Goh, W.S.S. Atlas of quantitative single-base-resolution N6-methyl-adenine methylomes. Nat. Commun. 2019, 10, 5636. [Google Scholar] [CrossRef]
- Nance, D.J.; Satterwhite, E.R.; Bhaskar, B.; Misra, S.; Carraway, K.R.; Mansfield, K.D. Characterization of METTL16 as a cytoplasmic RNA binding protein. PLoS ONE 2020, 15, e0227647. [Google Scholar] [CrossRef]
- Mikutis, S.; Gu, M.; Sendinc, E.; Hazemi, M.E.; Kiely-Collins, H.; Aspris, D.; Vassiliou, G.S.; Shi, Y.; Tzelepis, K.; Bernardes, G.J.L. meCLICK-Seq, a Substrate-Hijacking and RNA Degradation Strategy for the Study of RNA Methylation. ACS Cent. Sci. 2020, 6, 2196–2208. [Google Scholar] [CrossRef]
- Dai, Y.-Z.; Liu, Y.; Li, J.; Chen, M.-T.; Huang, M.; Wang, F.; Yang, Q.-S.; Yuan, J.-H.; Sun, S.-H. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B-AS1 in an m6A-dependent manner. Cell Mol. Biol. Lett. 2022, 27, 41. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Han, K.; Morgens, D.W.; Horii, T.; Kobayashi, R.; Tsuruyama, T.; Hia, F.; Yasukura, S.; Kajiya, A.; Cai, T.; et al. The N6-methyladenosine methyltransferase METTL16 enables erythropoiesis through safeguarding genome integrity. Nat. Commun. 2022, 13, 6435. [Google Scholar] [CrossRef]
- Shima, H.; Matsumoto, M.; Ishigami, Y.; Ebina, M.; Muto, A.; Sato, Y.; Kumagai, S.; Ochiai, K.; Suzuki, T.; Igarashi, K. S-Adenosylmethionine Synthesis Is Regulated by Selective N6-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep. 2017, 21, 3354–3363. [Google Scholar] [CrossRef]
- Satterwhite, E.R.; Mansfield, K.D. RNA methyltransferase METTL16: Targets and function. Wiley Interdiscip. Rev. RNA 2022, 13, e1681. [Google Scholar] [CrossRef]
- Mendel, M.; Delaney, K.; Pandey, R.R.; Chen, K.-M.; Wenda, J.M.; Vågbø, C.B.; Steiner, F.A.; Homolka, D.; Pillai, R.S. Splice site m6A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell 2021, 184, 3125–3142.e25. [Google Scholar] [CrossRef]
- Zeng, X.; Zhao, F.; Cui, G.; Zhang, Y.; Deshpande, R.A.; Chen, Y.; Deng, M.; Kloeber, J.A.; Shi, Y.; Zhou, Q.; et al. METTL16 antagonizes MRE11-mediated DNA end resection and confers synthetic lethality to PARP inhibition in pancreatic ductal adenocarcinoma. Nat. Cancer 2022, 3, 1088–1104. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, D.; Qu, J.; Zhuang, Y.; Peng, J.; Lan, S.; Zhang, S.; Huang, F. METTL16 predicts a favorable outcome and primes antitumor immunity in pancreatic ductal adenocarcinoma. Front. Cell Dev. Biol. 2022, 10, 759020. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Song, T.; Lv, S.; Li, N.; Zhao, X.; Ma, X.; Yan, Y.; Wang, W.; Sun, L. Versatile functions of RNA m6A machinery on chromatin. J. Mol. Cell Biol. 2022, 14, mjac011. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, V.V.; Jansen, P.W.T.C.; Baltissen, M.P.; Vermeulen, M.; Schneider, R. The interactome of a family of potential methyltransferases in HeLa cells. Sci. Rep. 2019, 9, 6584. [Google Scholar] [CrossRef] [PubMed]
- Covelo-Molares, H.; Obrdlik, A.; Poštulková, I.; Dohnálková, M.; Gregorová, P.; Ganji, R.; Potěšil, D.; Gawriyski, L.; Varjosalo, M.; Vaňáčová, Š. The comprehensive interactomes of human adenosine RNA methyltransferases and demethylases reveal distinct functional and regulatory features. Nucleic Acids Res. 2021, 49, 10895–10910. [Google Scholar] [CrossRef] [PubMed]
- Louloupi, A.; Ntini, E.; Conrad, T.; Ørom, U.A.V. Transient N-6-Methyladenosine Transcriptome Sequencing Reveals a Regulatory Role of m6A in Splicing Efficiency. Cell Rep. 2018, 23, 3429–3437. [Google Scholar] [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.A.F.O.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell 2017, 169, 326–337.e12. [Google Scholar] [CrossRef]
- Charenton, C.; Wilkinson, M.E.; Nagai, K. Mechanism of 5′ splice site transfer for human spliceosome activation. Science 2019, 364, 362–367. [Google Scholar] [CrossRef]
- Gu, J.; Patton, J.R.; Shimba, S.; Reddy, R. Localization of modified nucleotides in Schizosaccharomyces pombe spliceosomal small nuclear RNAs: Modified nucleotides are clustered in functionally important regions. RNA 1996, 2, 909–918. [Google Scholar]
- Wang, C.; Yang, J.; Song, P.; Zhang, W.; Lu, Q.; Yu, Q.; Jia, G. FIONA1 is an RNA N6-methyladenosine methyltransferase affecting Arabidopsis photomorphogenesis and flowering. Genome Biol. 2022, 23, 40. [Google Scholar] [CrossRef]
- Xu, T.; Wu, X.; Wong, C.E.; Fan, S.; Zhang, Y.; Zhang, S.; Liang, Z.; Yu, H.; Shen, L. FIONA1-Mediated m6 A Modification Regulates the Floral Transition in Arabidopsis. Adv. Sci. 2022, 9, e2103628. [Google Scholar] [CrossRef]
- Ishigami, Y.; Ohira, T.; Isokawa, Y.; Suzuki, Y.; Suzuki, T. A single m6A modification in U6 snRNA diversifies exon sequence at the 5′ splice site. Nat. Commun. 2021, 12, 3244. [Google Scholar] [CrossRef] [PubMed]
- Harada, F.; Kato, N.; Nishimura, S. The nucleotide sequence of nuclear 4.8S RNA of mouse cells. Biochem. Biophys. Res. Commun. 1980, 95, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Epstein, P.; Reddy, R.; Henning, D.; Busch, H. The nucleotide sequence of nuclear U6 (4.7 S) RNA. J. Biol. Chem. 1980, 255, 8901–8906. [Google Scholar] [CrossRef]
- Didychuk, A.L.; Butcher, S.E.; Brow, D.A. The life of U6 small nuclear RNA, from cradle to grave. RNA 2018, 24, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Madhani, H.D.; Bordonné, R.; Guthrie, C. Multiple roles for U6 snRNA in the splicing pathway. Genes Dev. 1990, 4, 2264–2277. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, G.L. Biological methylation: Selected aspects. Annu. Rev. Biochem. 1975, 44, 435–451. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef]
- Lauinger, L.; Kaiser, P. Sensing and Signaling of Methionine Metabolism. Metabolites 2021, 11, 83. [Google Scholar] [CrossRef]
- Vázquez-Chantada, M.; Fernández-Ramos, D.; Embade, N.; Martínez-Lopez, N.; Varela-Rey, M.; Woodhoo, A.; Luka, Z.; Wagner, C.; Anglim, P.P.; Finnell, R.H.; et al. HuR/methyl-HuR and AUF1 regulate the MAT expressed during liver proliferation, differentiation, and carcinogenesis. Gastroenterology 2010, 138, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Ramani, K.; Mato, J.M.; Lu, S.C. Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers 2011, 3, 1480–1497. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.; Barbier-Torres, L.; Fan, W.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferases in liver cancer. World J. Gastroenterol. 2019, 25, 4300–4319. [Google Scholar] [CrossRef]
- Kotb, M.; Mudd, S.H.; Mato, J.M.; Geller, A.M.; Kredich, N.M.; Chou, J.Y.; Cantoni, G.L. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997, 13, 51–52. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; Latasa, M.U.; Varela-Rey, M.; Lu, S.C.; García-Trevijano, E.R.; Mato, J.M.; Avila, M.A. L-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: Role of S-adenosylmethionine. J. Biol. Chem. 2003, 278, 19885–19890. [Google Scholar] [CrossRef]
- Bresson, S.M.; Hunter, O.V.; Hunter, A.C.; Conrad, N.K. Canonical Poly(A) Polymerase Activity Promotes the Decay of a Wide Variety of Mammalian Nuclear RNAs. PLoS Genet. 2015, 11, e1005610. [Google Scholar] [CrossRef]
- Yang, H.; Huang, Z.Z.; Zeng, Z.; Chen, C.; Selby, R.R.; Lu, S.C. Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G184–G190. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; García-Trevijano, E.R.; Latasa, M.; Martín-Duce, A.; Fortes, P.; Caballería, J.; Avila, M.A.; Mato, J.M. Methionine adenosyltransferase II β subunit gene expression provides a proliferative advantage in human hepatoma. Gastroenterology 2003, 124, 940–948. [Google Scholar] [CrossRef]
- Batey, R.T. Recognition of S-adenosylmethionine by riboswitches. Wiley Interdiscip. Rev. RNA 2011, 2, 299–311. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, W.; Chen, D.; Murchie, A.I.H. Interactions between SAM and the 5′ UTR mRNA of the sam1 gene regulate translation in S. pombe. RNA 2020, 26, 150–161. [Google Scholar] [CrossRef]
- Breaker, R.R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4, a003566. [Google Scholar] [CrossRef]
- Winkler, W.C.; Nahvi, A.; Sudarsan, N.; Barrick, J.E.; Breaker, R.R. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol. 2003, 10, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Moltke, I.; Roth, A.; Washietl, S.; Wen, J.; Kellis, M.; Breaker, R.; Pedersen, J.S. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 2011, 21, 1929–1943. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Park, S.-K.; Hunter, O.V.; Bresson, S.M.; Conrad, N.K. Balance between MAT2A intron detention and splicing is determined cotranscriptionally. RNA 2018, 24, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, A.M.; Flaherty, J.N.; Hunter, O.V.; Liu, K.; Kumar, A.; Xing, C.; Tu, B.P.; Conrad, N.K. SAM homeostasis is regulated by CFIm-mediated splicing of MAT2A. eLife 2021, 10, e64930. [Google Scholar] [CrossRef] [PubMed]
- Masamha, C.P. The emerging roles of CFIm25 (NUDT21/CPSF5) in human biology and disease. Wiley Interdiscip. Rev. RNA 2022, e1757. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kang, J.; Su, J.; Zhang, J.; Zhang, L.; Liu, X.; Zhang, J.; Wang, F.; Lu, Z.; Xing, X.; et al. Methionine adenosyltransferase 2A regulates mouse zygotic genome activation and morula to blastocyst transition†. Biol. Reprod. 2019, 100, 601–617. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Watabe, E.; Togo-Ohno, M.; Ishigami, Y.; Wani, S.; Hirota, K.; Kimura-Asami, M.; Hasan, S.; Takei, S.; Fukamizu, A.; Suzuki, Y.; et al. m6 A-mediated alternative splicing coupled with nonsense-mediated mRNA decay regulates SAM synthetase homeostasis. EMBO J. 2021, 40, e106434. [Google Scholar] [CrossRef]
- Sendinc, E.; Valle-Garcia, D.; Jiao, A.; Shi, Y. Analysis of m6A RNA methylation in Caenorhabditis elegans. Cell Discov. 2020, 6, 47. [Google Scholar] [CrossRef]
- Yoshida, H.; Park, S.-Y.; Sakashita, G.; Nariai, Y.; Kuwasako, K.; Muto, Y.; Urano, T.; Obayashi, E. Elucidation of the aberrant 3′ splice site selection by cancer-associated mutations on the U2AF1. Nat. Commun. 2020, 11, 4744. [Google Scholar] [CrossRef]
- Towbin, B.D.; González-Aguilera, C.; Sack, R.; Gaidatzis, D.; Kalck, V.; Meister, P.; Askjaer, P.; Gasser, S.M. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 2012, 150, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.; Yeom, M.; Kim, J.-H.; Nam, H.G. FIONA1 is essential for regulating period length in the Arabidopsis circadian clock. Plant Cell 2008, 20, 307–319. [Google Scholar] [CrossRef]
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m6 A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef]
- Bodi, Z.; Zhong, S.; Mehra, S.; Song, J.; Graham, N.; Li, H.; May, S.; Fray, R.G. Adenosine Methylation in Arabidopsis mRNA is Associated with the 3′ End and Reduced Levels Cause Developmental Defects. Front. Plant Sci. 2012, 3, 48. [Google Scholar] [CrossRef]
- Li, W.; Han, Y.; Tao, F.; Chong, K. Knockdown of SAMS genes encoding S-adenosyl-l-methionine synthetases causes methylation alterations of DNAs and histones and leads to late flowering in rice. J. Plant Physiol. 2011, 168, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Sekula, B.; Ruszkowski, M.; Dauter, Z. S-adenosylmethionine synthases in plants: Structural characterization of type I and II isoenzymes from Arabidopsis thaliana and Medicago truncatula. Int. J. Biol. Macromol. 2020, 151, 554–565. [Google Scholar] [CrossRef]
- Wang, X.-K.; Zhang, Y.-W.; Wang, C.-M.; Li, B.; Zhang, T.-Z.; Zhou, W.-J.; Cheng, L.-J.; Huo, M.-Y.; Zhang, C.-H.; He, Y.-L. METTL16 promotes cell proliferation by up-regulating cyclin D1 expression in gastric cancer. J. Cell Mol. Med. 2021, 25, 6602–6617. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Ward, R.L. Folate and one-carbon metabolism and its impact on aberrant DNA methylation in cancer. Adv. Genet. 2010, 71, 79–121. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Altundag, Ö.; Canpinar, H.; Çelebi-Saltik, B. Methionine affects the expression of pluripotency genes and protein levels associated with methionine metabolism in adult, fetal, and cancer stem cells. J. Cell Biochem. 2022, 123, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.-K.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef]
- Sharma, U.; Rando, O.J. Metabolic Inputs into the Epigenome. Cell Metab. 2017, 25, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Serefidou, M.; Venkatasubramani, A.V.; Imhof, A. The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet. 2019, 10, 764. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).