
Deviations from Mendelian Inheritance on Bovine X-Chromosome Revealing Recombination, Sex-of-Offspring Effects and Fertility-Related Candidate Genes
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genotype Data
2.2. Imputation
2.3. Male- and Female-Offspring Specific-TRD
2.4. Statistical Analyses
2.5. Pseudoautosomal Characterization
2.6. Functional Analyses
3. Results and Discussion
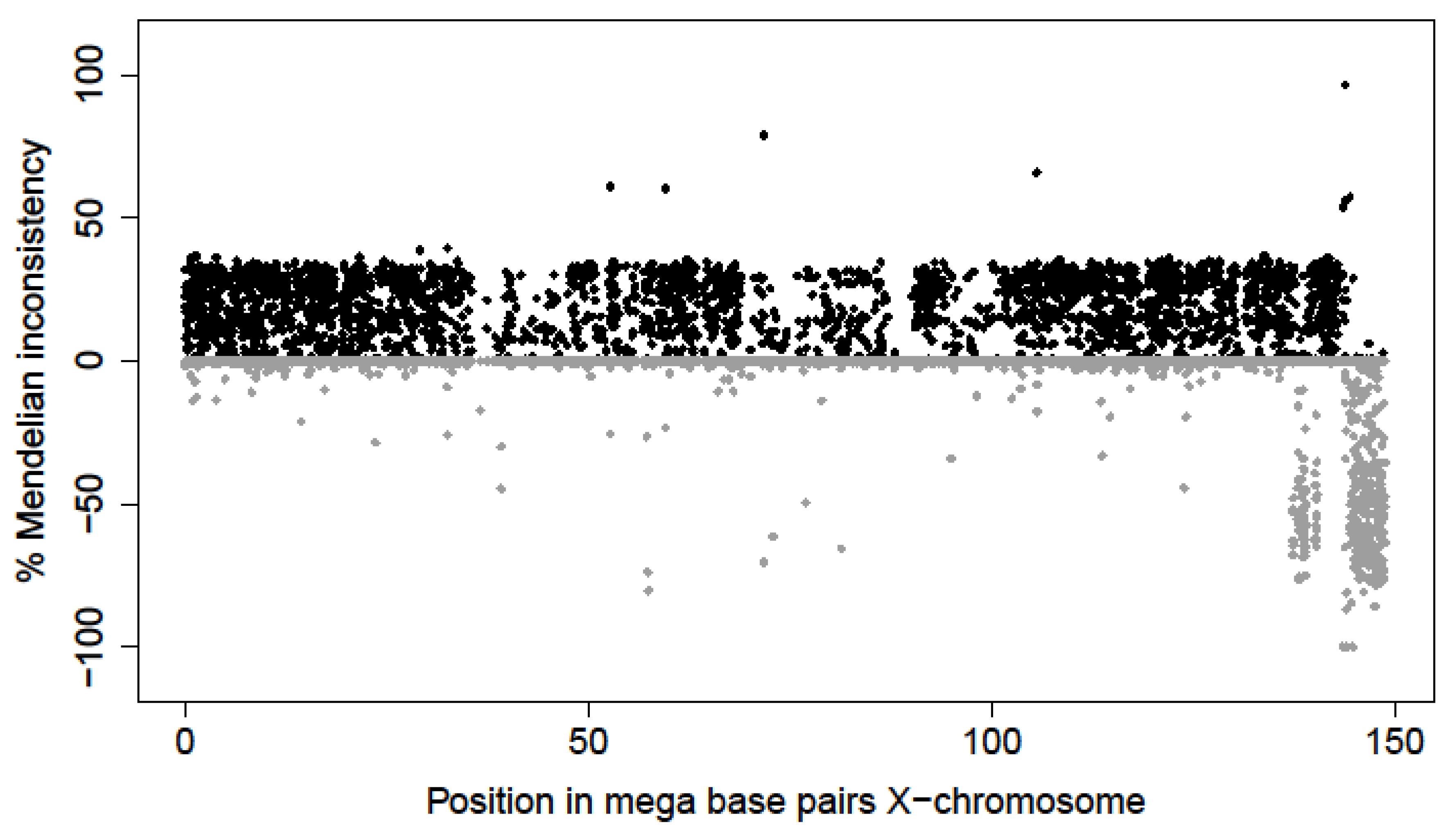
3.1. Characterization of X-Chromosome Using Parent-Offspring Genotyped Trios
3.2. Deviations from Mendelian Inheritance on the Pseudoautosomal Part of the Sex Chromosomes
3.3. Opposite Sire-TRD between Male- and Female-Offspring
3.4. Specific Sex-Offspring Sire-TRD
3.5. Deviations from Mendelian Inheritance on the Heterosomal Part of the X-Chromosome
3.5.1. SNP-by-SNP Analysis
3.5.2. Haplotype Analysis
3.6. Recessive TRD Pattern
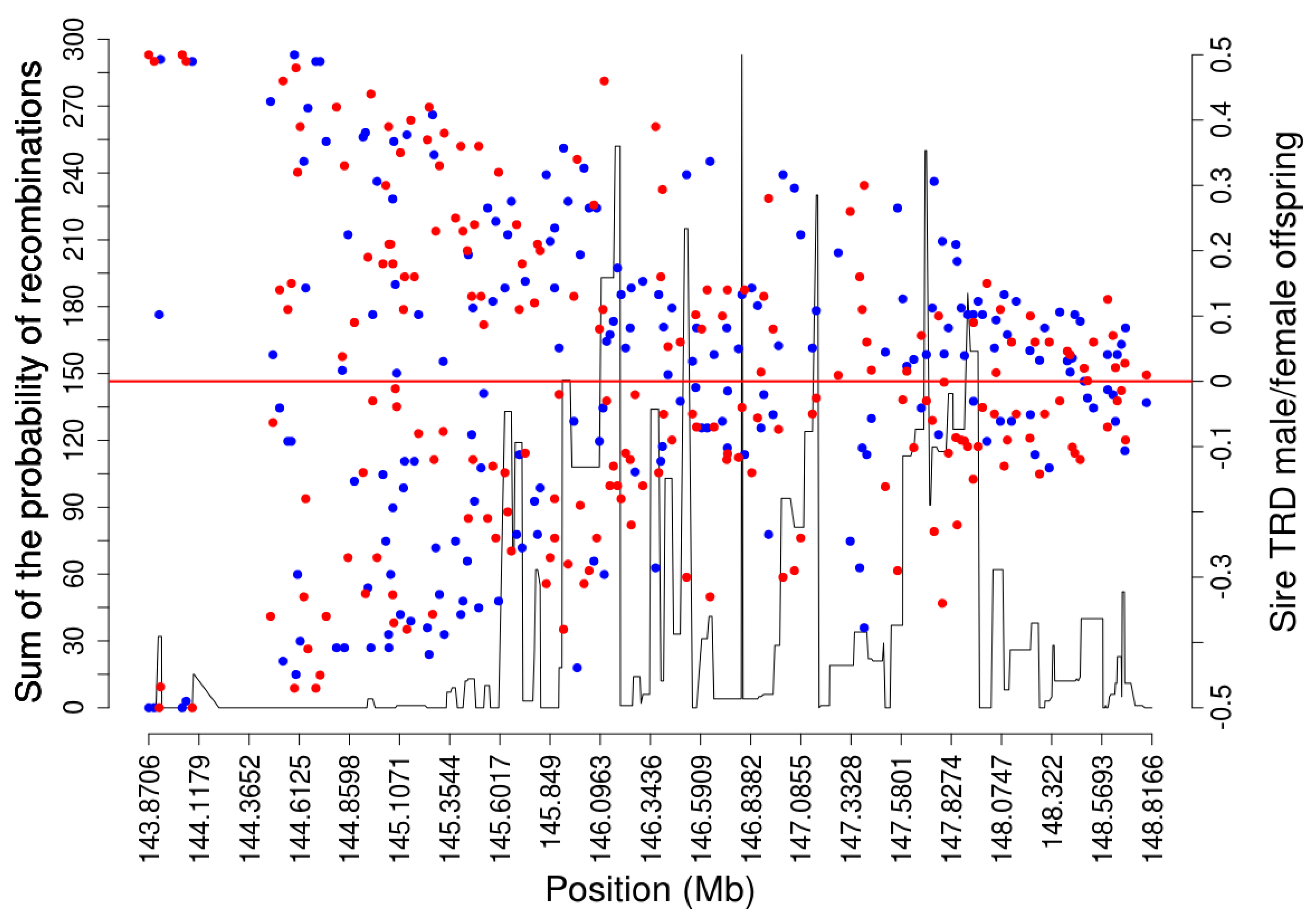
3.7. Functional Impact around the Pseudoautosomal Boundaries Identified Using Opposite TRD Patterns between Male and Female Offspring
3.8. Enriched Metabolic Pathways, Diseases and Biological Functions Associated with the Positional Candidate Genes Displaying Deviations from Mendelian Inheritance on the X-Chromosome
3.9. Functional Candidate Genes Identified in the Candidate Regions with Deviations from Mendelian Expectations
3.10. Validation of Functional Candidate Genes Previously Identified in Independent Populations Using Genome-Wide Association Studies’ Meta-Analysis for Fertility Traits in Cattle
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dunn, L.C. Evidence of evolutionary forces leading to the spread of lethal genes in wild populations of house mice. Proc. Natl. Acad. Sci. USA 1957, 43, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatib, H.; Monson, R.L.; Schutzkus, V.; Kohl, D.M.; Rosa, G.J.M.; Rutledge, J.J. Mutations in the STAT5A Gene Are Associated with Embryonic Survival and Milk Composition in Cattle. J. Dairy Sci. 2008, 91, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casellas, J.; Gularte, R.J.; Farber, C.R.; Varona, L.; Mehrabian, M.; Schadt, E.E.; Lusis, A.J.; Attie, A.D.; Yandell, B.S.; Medrano, J.F. Genome Scans for Transmission Ratio Distortion Regions in Mice. Genetics 2012, 191, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casellas, J.; Cañas-Álvarez, J.J.; González-Rodríguez, A.; Puig-Oliveras, A.; Fina, M.; Piedrafita, J.; Molina, A.; Díaz, C.; Baró, J.A.; Varona, L. Bayesian Analysis of Parent-Specific Transmission Ratio Distortion in Seven Spanish Beef Cattle Breeds. Anim. Genet. 2017, 48, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, E.A.; Id-Lahoucine, S.; Cánovas, A.; Casellas, J.; Schenkel, F.S.; Wood, B.J.; Baes, C.F. Discovering Lethal Alleles across the Turkey Genome Using a Transmission Ratio Distortion Approach. Anim. Genet. 2020, 51, 876–889. [Google Scholar] [CrossRef]
- Huang, L.O.; Labbe, A.; Infante-Rivard, C. Transmission Ratio Distortion: Review of Concept and Implications for Genetic Association Studies. Hum. Genet. 2013, 132, 245–263. [Google Scholar] [CrossRef]
- Fishman, L.; Willis, J.H. A Novel Meiotic Drive Locus Almost Completely Distorts Segregation in Mimulus (Monkeyflower) Hybrids. Genetics 2005. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M.; Olson, K.M.; Null, D.J.; Hutchison, J.L. Harmful Recessive Effects on Fertility Detected by Absence of Homozygous Haplotypes. J. Dairy Sci. 2011, 94, 6153–6161. [Google Scholar] [CrossRef] [Green Version]
- Casellas, J.; Manunza, A.; Mercader, A.; Quintanilla, R.; Amills, M. A Flexible Bayesian Model for Testing for Transmission Ratio Distortion. Genetics 2014, 198, 1357–1367. [Google Scholar] [CrossRef]
- Hoff, J.L.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F. Candidate Lethal Haplotypes and Causal Mutations in Angus Cattle. BMC Genom. 2017, 18, 799. [Google Scholar] [CrossRef]
- Howard, D.M.; Pong-Wong, R.; Knap, P.W.; Woolliams, J.A. Use of Haplotypes to Identify Regions Harbouring Lethal Recessive Variants in Pigs. Genet. Sel. Evol. 2017, 49, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Id-Lahoucine, S.; Cánovas, A.; Jaton, C.; Miglior, F.; Fonseca, P.A.S.; Sargolzaei, M.; Miller, S.; Schenkel, F.S.; Medrano, J.F.; Casellas, J. Implementation of Bayesian Methods to Identify SNP and Haplotype Regions with Transmission Ratio Distortion across the Whole Genome: TRDscan v.1.0. J. Dairy Sci. 2019, 102, 3175–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumova, A.K.; Leppert, M.; Barker, D.F.; Morgan, K.; Sapienza, C. Parental Origin-Dependent, Male Offspring-Specific Transmission-Ratio Distortion at Loci on the Human X Chromosome. Am. J. Hum. Genet. 1998, 102, 3175–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumova, A.K.; Olien, L.; Bird, L.M.; Slamka, C.; Fonseca, M.; Verner, A.E.; Wang, M.; Leppert, M.; Morgan, K.; Sapienza, C. Transmission-ratio Distortion of X Chromosomes among Male Offspring of Females with Skewed X-inactivation. Dev. Genet. 1995, 62, 1493–1499. [Google Scholar] [CrossRef]
- Id-Lahoucine, S.; Casellas, J. Impact of Incomplete Pedigree Data and Independent Culling Level Pre-Selection on the Genetic Evaluation of Livestock: A Simulation Study on Lamb Growth. Livest. Sci. 2017, 198, 76–81. [Google Scholar] [CrossRef]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 198, 76–81. [Google Scholar] [CrossRef]
- Ferdosi, M.H.; Kinghorn, B.P.; van der Werf, J.H.J.; Lee, S.H.; Gondro, C. Hsphase: An R Package for Pedigree Reconstruction, Detection of Recombination Events, Phasing and Imputation of Half-Sib Family Groups. BMC Bioinform. 2014, 90, 773–795. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R Package for Genomic Annotation and Integration of Multiple Data Sources in Livestock for Positional Candidate Loci. Gigascience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/ Bioconductor Package BiomaRt. Nat. Protoc. 2009, 9, giaa149. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, T.F.; Cánovas, A.; Canela-Xandri, O.; González-Prendes, R.; Amills, M.; Quintanilla, R. RNA-Seq Based Detection of Differentially Expressed Genes in the Skeletal Muscle of Duroc Pigs with Distinct Lipid Profiles. Sci. Rep. 2017, 4, 1184–1191. [Google Scholar] [CrossRef]
- Das, P.J.; Chowdhary, B.P.; Raudsepp, T. Characterization of the Bovine Pseudoautosomal Region and Comparison with Sheep, Goat, and Other Mammalian Pseudoautosomal Regions. Cytogenet. Genome Res. 2009, 126, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Couldrey, C.; Johnson, T.; Lopdell, T.; Zhang, I.L.; Littlejohn, M.D.; Keehan, M.; Sherlock, R.G.; Tiplady, K.; Scott, A.; Davis, S.R.; et al. Bovine Mammary Gland X Chromosome Inactivation. J. Dairy Sci. 2017, 100, 5491–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.; Keehan, M.; Harland, C.; Lopdell, T.; Spelman, R.J.; Davis, S.R.; Rosen, B.D.; Smith, T.P.L.; Couldrey, C. Short Communication: Identification of the Pseudoautosomal Region in the Hereford Bovine Reference Genome Assembly ARS-UCD1.2. J. Dairy Sci. 2019, 102, 3254–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudsepp, T.; Das, P.J.; Avila, F.; Chowdhary, B.P. The Pseudoautosomal Region and Sex Chromosome Aneuploidies in Domestic Species. Sex. Dev. 2012, 6, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N. Recombination, GC-Content and the Human Pseudoautosomal Boundary Paradox. Trends Genet. 2004, 20, 347–349. [Google Scholar] [CrossRef] [PubMed]
- McClure, M.C.; McCarthy, J.; Flynn, P.; McClure, J.C.; Dair, E.; O’Connell, D.K.; Kearney, J.F. SNP Data Quality Control in a National Beef and Dairy Cattle System and Highly Accurate SNP Based Parentage Verification and Identification. Front. Genet. 2018, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Jeffreys, H. Theory of Probability. Philos. Sci. 1940, 7, 263–264. [Google Scholar] [CrossRef]
- Rouyer, F.; Simmler, M.C.; Johnsson, C.; Vergnaud, G.; Cooke, H.J.; Weissenbach, J. A Gradient of Sex Linkage in the Pseudoautosomal Region of the Human Sex Chromosomes. Nature 1986, 319, 291–295. [Google Scholar] [CrossRef]
- Lien, S.; Szyda, J.; Schechinger, B.; Rappold, G.; Arnheim, N. Evidence for Heterogeneity in Recombination in the Human Pseudoautosomal Region: High Resolution Analysis by Sperm Typing and Radiation-Hybrid Mapping. Am. J. Hum. Genet. 2000, 66, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Hinch, A.G.; Altemose, N.; Noor, N.; Donnelly, P.; Myers, S.R. Recombination in the Human Pseudoautosomal Region PAR1. PLoS Genet. 2014, 10, e1004503. [Google Scholar] [CrossRef]
- Naumova, A.; Sapienza, C. The Genetics of Retinoblastoma, Revisited. Am. J. Hum. Genet. 1994, 54, 264–273. [Google Scholar] [PubMed]
- Bourc’His, D.; Bestor, T.H. Origins of Extreme Sexual Dimorphism in Genomic Imprinting. Cytogenet. Genome Res. 2006, 113, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.M. Sex Chromosome Specialization and Degeneration in Mammals. Cell 2006, 124, 901–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casellas, J.; Id-Lahoucine, S.; Cánovas, A. Discriminating between Allele- and Genotype-Specific Transmission Ratio Distortion. Anim. Genet. 2020, 51, 847–854. [Google Scholar] [CrossRef]
- Van Laere, A.S.; Coppieters, W.; Georges, M. Characterization of the Bovine Pseudoautosomal Boundary: Documenting the Evolutionary History of Mammalian Sex Chromosomes. Genome Res. 2008, 18, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ikeguchi, Y.; McCloskey, D.E.; Nelson, P.; Pegg, A.E. Spermine Synthesis Is Required for Normal Viability, Growth, and Fertility in the Mouse. J. Biol. Chem. 2004, 49, 51370–51375. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Aoyama, C.; Young, S.G.; Vance, D.E. Early Embryonic Lethality Caused by Disruption of the Gene for Choline Kinase α, the First Enzyme in Phosphatidylcholine Biosynthesis. J. Biol. Chem. 2008, 279, 51370–51375. [Google Scholar] [CrossRef] [Green Version]
- Gunter, C.; Frank, M.; Tian, Y.; Murti, K.G.; Rehg, J.E.; Jackowski, S. Probucol Therapy Overcomes the Reproductive Defect in CTP: Phosphocholine Cytidylyltransferase Β2 Knockout Mice. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 283, 1456–1462. [Google Scholar] [CrossRef] [Green Version]
- Anagnostopoulos, A.V. It’s a Knockout! Male Infertility and Neuropathology. Trends Genet. 2002, 18, 8–10. [Google Scholar] [CrossRef]
- Cariboni, A.; Maggi, R.; Parnavelas, J.G. From Nose to Fertility: The Long Migratory Journey of Gonadotropin-Releasing Hormone Neurons. Trends Neurosci. 2007, 30, 638–644. [Google Scholar] [CrossRef]
- Fullerton, G.; Hamilton, M.; Maheshwari, A. Should Non-Mosaic Klinefelter Syndrome Men Be Labelled as Infertile in 2009? Hum. Reprod. 2010, 25, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killeen, A.P.; Morris, D.G.; Kenny, D.A.; Mullen, M.P.; Diskin, M.G.; Waters, S.M. Global Gene Expression in Endometrium of High and Low Fertility Heifers during the Mid-Luteal Phase of the Estrous Cycle. BMC Genom. 2014, 15, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.J.; Gropman, A.L.; Sapp, J.C.; Teer, J.K.; Martin, J.M.; Liu, C.F.; Yuan, X.; Ye, Z.; Cheng, L.; Brodsky, R.A.; et al. The Phenotype of a Germline Mutation in PIGA: The Gene Somatically Mutated in Paroxysmal Nocturnal Hemoglobinuria. Am. J. Hum. Genet. 2012, 90, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawagoe, K.; Kitamura, D.; Okabe, M.; Taniuchi, I.; Ikawa, M.; Watanabe, T.; Kinoshita, T.; Takeda, J. Glycosylphosphatidylinositol-Anchor-Deficient Mice: Implications for Clonal Dominance of Mutant Cells in Paroxysmal Nocturnal Hemoglobinuria. Blood 1996, 87, 3600–3606. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; An, P.; Zhang, R.; He, X.; Yin, G.; Min, W. Etk/Bmx as a Tumor Necrosis Factor Receptor Type 2-Specific Kinase: Role in Endothelial Cell Migration and Angiogenesis. Mol. Cell. Biol. 2002, 22, 7512–7523. [Google Scholar] [CrossRef] [Green Version]
- Stoletov, K.V.; Terman, B.I. Bmx Is a Downstream Rap1 Effector in VEGF-Induced Endothelial Cell Activation. Biochem. Biophys. Res. Commun. 2004, 320, 70–75. [Google Scholar] [CrossRef]
- Cui, Y.; Yan, H.; Wang, K.; Xu, H.; Zhang, X.; Zhu, H.; Liu, J.; Qu, L.; Lan, X.; Pan, C. Insertion/Deletion within the KDM6A Gene Is Significantly Associated with Litter Size in Goat. Front. Genet. 2018, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Puigvert, J.C.; Von Stechow, L.; Siddappa, R.; Pines, A.; Bahjat, M.; Haazen, L.C.J.M.; Olsen, J.V.; Vrieling, H.; Meerman, J.H.N.; Mullenders, L.H.F.; et al. Systems Biology Approach Identifies the Kinase Csnk1a1 as a Regulator of the DNA Damage Response in Embryonic Stem Cells. Sci. Signal. 2013, 6, ra5. [Google Scholar] [CrossRef] [Green Version]
- Feng, R.; Sang, Q.; Zhu, Y.; Fu, W.; Liu, M.; Xu, Y.; Shi, H.; Xu, Y.; Qu, R.; Chai, R.; et al. MiRNA-320 in the Human Follicular Fluid Is Associated with Embryo Quality in Vivo and Affects Mouse Embryonic Development in Vitro. Sci. Rep. 2015, 5, 8689. [Google Scholar] [CrossRef]
- Song, M.; Yang, H.; Yao, S.; Ma, F.; Li, Z.; Deng, Y.; Deng, H.; Zhou, Q.; Lin, S.; Wei, Y. A Critical Role of Vascular Endothelial Growth Factor D in Zebrafish Embryonic Vasculogenesis and Angiogenesis. Biochem. Biophys. Res. Commun. 2007, 357, 924–930. [Google Scholar] [CrossRef]
- Nicoli, S.; Knyphausen, C.P.; Zhu, L.J.; Lakshmanan, A.; Lawson, N.D. MiR-221 Is Required for Endothelial Tip Cell Behaviors during Vascular Development. Dev. Cell 2012, 22, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Chen, M.H.; Moskowitz, I.P.; Mendonza, A.M.; Vidali, L.; Nakamura, F.; Kwiatkowski, D.J.; Walsh, C.A. Filamin a (FLNA) Is Required for Cell-Cell Contact in Vascular Development and Cardiac Morphogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19836–19841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puranam, R.S.; He, X.P.; Yao, L.; Le, T.; Jang, W.; Rehder, C.W.; Lewis, D.V.; McNamara, J.O. Disruption of Fgf13 Causes Synaptic Excitatory–Inhibitory Imbalance and Genetic Epilepsy and Febrile Seizures Plus. J. Neurosci. 2015, 35, 8866–8881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, P.; Skarnes, W.; Bird, A. The Methyl-CpG Binding Protein MeCP2 Is Essential for Embryonic Development in the Mouse. Nat. Genet. 1996, 12, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, M.; Deogracias, R.; Guy, J.; Poot, R.A.; Bird, A.; Barde, Y.A. Disease Modeling Using Embryonic Stem Cells: MeCP2 Regulates Nuclear Size and RNA Synthesis in Neurons. Stem Cells 2012, 12, 205–208. [Google Scholar] [CrossRef]
- Docheva, D.; Hunziker, E.B.; Fässler, R.; Brandau, O. Tenomodulin Is Necessary for Tenocyte Proliferation and Tendon Maturation. Mol. Cell Biol. 2005, 30, 2128–2139. [Google Scholar] [CrossRef] [Green Version]
- Naruse, M.; Ono, R.; Irie, M.; Nakamura, K.; Furuse, T.; Hino, T.; Oda, K.; Kashimura, M.; Yamada, I.; Wakana, S.; et al. Sirh7/Ldoc1 Knockout Mice Exhibit Placental P4 Overproduction and Delsayed Parturition. Development 2014, 141, 4763–4771. [Google Scholar] [CrossRef] [Green Version]
- Suarez, P.E.; Rodriguez, E.G.; Soundararajan, R.; Mérillat, A.M.; Stehle, J.C.; Rotman, S.; Roger, T.; Voirol, M.J.; Wang, J.; Gross, O.; et al. The Glucocorticoid-Induced Leucine Zipper (Gilz/Tsc22d3-2) Gene Locus Plays a Crucial Role in Male Fertility. Mol. Endocrinol. 2012, 26, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- De Souza Fonseca, P.A.; dos Santos, F.C.; Lam, S.; Suárez-Vega, A.; Miglior, F.; Schenkel, F.S.; de Diniz, L.A.F.; Id-Lahoucine, S.; Carvalho, M.R.S.; Cánovas, A. Genetic Mechanisms Underlying Spermatic and Testicular Traits within and among Cattle Breeds: Systematic Review and Prioritization of GWAS Results. J. Anim. Sci. 2018, 96, 4978–4999. [Google Scholar] [CrossRef]
- Dunn, H.G.; MacLeod, P.M. Rett Syndrome: Review of Biological Abnormalities. Can. J. Neurol. Sci. 2001, 28, 16–29. [Google Scholar] [CrossRef]
- Sanmann, J.N.; Bishay, D.L.; Starr, L.J.; Bell, C.A.; Pickering, D.L.; Stevens, J.M.; Kahler, S.G.; Olney, A.H.; Schaefer, G.B.; Sanger, W.G. Characterization of Six Novel Patients with MECP2 Duplications Due to Unbalanced Rearrangements of the X Chromosome. Am. J. Med. Genet. A 2012, 158, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Tsuji-Hosokawa, A.; Matsuda, N.; Kurosawa, K.; Kashimada, K.; Morio, T. A Case of MECP2 Duplication Syndrome with Gonadotropin-Dependent Precocious Puberty. Horm. Res. Paediatr. 2017, 87, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Finas, D.; Huszar, M.; Agic, A.; Dogan, S.; Kiefel, H.; Riedle, S.; Gast, D.; Marcovich, R.; Noack, F.; Altevogt, P.; et al. L1 Cell Adhesion Molecule (L1CAM) as a Pathogenetic Factor in Endometriosis. Hum. Reprod. 2008, 23, 1053–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiess, A.N.; Feig, C.; Schulze, W.; Chalmel, F.; Cappallo-Obermann, H.; Primig, M.; Kirchhoff, C. Cross-Platform Gene Expression Signature of Human Spermatogenic Failure Reveals Inflammatory-like Response. Hum. Reprod. 2007, 22, 2936–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, H.; Skiniotis, G.; Spilker, A.; Kandels-Lewis, S.; Vernos, I.; Surrey, T. A Kinesin-like Motor Inhibits Microtubule Dynamic Instability. Science 2004, 303, 1519–1522. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Pan, M.H.; Lu, Y.; Wan, X.; Zhang, Y.; Sun, S.C. Involvement of Kif4a in Spindle Formation and Chromosome Segregation in Mouse Oocytes. Aging Dis. 2018, 9, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Neupane, M.; Geary, T.W.; Kiser, J.N.; Burns, G.W.; Hansen, P.J.; Spencer, T.E.; Neibergs, H.L. Loci and Pathways Associated with Uterine Capacity for Pregnancy and Fertility in Beef Cattle. PLoS ONE 2017, 12, e0188997. [Google Scholar] [CrossRef] [Green Version]
- Jansen, E.J.R.; Martens, G.J.M. Novel Insights into V-ATPase Functioning: Distinct Roles for Its Accessory Subunits ATP6AP1/Ac45 and ATP6AP2/(pro) Renin Receptor. Curr. Protein Pept. Sci. 2012, 13, 124–133. [Google Scholar] [CrossRef]
- Jansen, E.J.R.; Timal, S.; Ryan, M.; Ashikov, A.; Van Scherpenzeel, M.; Graham, L.A.; Mandel, H.; Hoischen, A.; Iancu, T.C.; Raymond, K.; et al. ATP6AP1 Deficiency Causes an Immunodeficiency with Hepatopathy, Cognitive Impairment and Abnormal Protein Glycosylation. Nat. Commun. 2016, 13, 124–133. [Google Scholar] [CrossRef]
- Gokey, J.J.; Dasgupta, A.; Amack, J.D. The V-ATPase Accessory Protein Atp6ap1b Mediates Dorsal Forerunner Cell Proliferation and Left-Right Asymmetry in Zebrafish. Dev. Biol. 2015, 7, 11600. [Google Scholar] [CrossRef]
- Gloria-Bottini, F.; Meloni, G.F.; MacMurray, J.P.; Cosmi, E.; Meloni, T.; Bottini, E. Evidence of Decreased Fertility in Women Carrying the Gene for G6PD Deficiency: A Study in the Sardinian Population. In The Biodemography of Human Reproduction and Fertility; Springer: Cham, Switzerland, 2003. [Google Scholar]
- Lian, G.; Lu, J.; Hu, J.; Zhang, J.; Cross, S.H.; Ferland, R.J.; Sheen, V.L. Filamin a Regulates Neural Progenitor Proliferation and Cortical Size through Wee1-Dependent Cdk1 Phosphorylation. J. Neurosci. 2012, 32, 7672–7684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, L.A.; Nguyen, L.S.; Domingo, D.; Sun, Y.; Barry, S.; Hancarova, M.; Plevova, P.; Vlckova, M.; Havlovicova, M.; Kalscheuer, V.M.; et al. HCFC1 Loss-of-Function Mutations Disrupt Neuronal and Neural Progenitor Cells of the Developing Brain. Hum. Mol. Genet. 2015, 24, 3335–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tüttelmann, F.; Simoni, M.; Kliesch, S.; Ledig, S.; Dworniczak, B.; Wieacker, P.; Röpke, A. Copy Number Variants in Patients with Severe Oligozoospermia and Sertoli-Cell-Only Syndrome. PLoS ONE 2011, 6, e19426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatsenko, A.N.; Georgiadis, A.P.; Röpke, A.; Berman, A.J.; Jaffe, T.; Olszewska, M.; Westernströer, B.; Sanfilippo, J.; Kurpisz, M.; Rajkovic, A.; et al. X-Linked TEX11 Mutations, Meiotic Arrest, and Azoospermia in Infertile Men. N. Engl. J. Med. 2015, 372, 2097–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Camargo, G.M.F.; Porto-Neto, L.R.; Kelly, M.J.; Bunch, R.J.; McWilliam, S.M.; Tonhati, H.; Lehnert, S.A.; Fortes, M.R.S.; Moore, S.S. Non-Synonymous Mutations Mapped to Chromosome X Associated with Andrological and Growth Traits in Beef Cattle. BMC Genom. 2015, 16, 384. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordinates in Base Pairs | Frequency | Male-Offspring | Female-Offspring | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n° Hetero 1 Sires | n° Hetero Dams | n° Info 2 Offspring | Sire-TRD (αs♂) | BF | n° Hetero Sires | n° Hetero Dams | n° Info Offspring | Sire-TRD (αs♀) | BF | ||
| 137,464,119 | 0.63 | 376 | 2307 | 10,297 | 0.02 | 0.79 | 275 | 1625 | 7100 | −0.04 | 104.2 |
| 138,080,118 | 0.46 | 699 | 4368 | 19,985 | −0.01 | 0.04 | 1281 | 20,752 | 48,445 | 0.03 | 1022.8 |
| 144,824,704 | 0.31 | 267 | 1691 | 8037 | 0.01 | 0.03 | 365 | 2258 | 8646 | 0.04 | 103.4 |
| 144,927,188 | 0.83 | 7 | 88 | 228 | 0.36 | 103.8 | 8 | 46 | 107 | −0.14 | 0.63 |
| 145,093,201 | 0.24 | 403 | 2284 | 10,759 | 0.00 | 0.02 | 361 | 1879 | 9007 | −0.04 | 104.2 |
| 145,322,565 | 0.67 | 280 | 1532 | 9045 | 0.02 | 1 | 364 | 1946 | 9432 | −0.08 | 1027.4 |
| 146,268,179 | 0.14 | 216 | 1351 | 6317 | −0.15 | 1058.9 | 334 | 5575 | 13,299 | −0.02 | 3.98 |
| 146,403,456 | 0.08 | 14 | 51 | 155 | −0.11 | 0.79 | 13 | 19 | 115 | 0.29 | 105.9 |
| 147,269,713 | 0.85 | 56 | 162 | 809 | 0.18 | 109.2 | 33 | 132 | 442 | 0.01 | 0.08 |
| 147,433,504 | 0.52 | 302 | 2111 | 10,749 | −0.07 | 1018.8 | 406 | 2793 | 11,668 | 0.02 | 0.63 |
| 147,587,823 | 0.58 | 98 | 290 | 1456 | 0.11 | 108.3 | 66 | 209 | 764 | −0.03 | 0.13 |
| 148,398,942 | 0.22 | 477 | 2871 | 14,803 | 0.02 | 10 | 851 | 14,732 | 37,427 | 0.05 | 1031.7 |
| 148,498,697 | 0.34 | 267 | 1685 | 8671 | −0.04 | 102.9 | 338 | 2173 | 8922 | 0.00 | 0.02 |
| 148,665,523 | 0.61 | 269 | 1853 | 8777 | 0.05 | 105.4 | 351 | 2335 | 8873 | −0.01 | 0.10 |
| 148,682,952 | 0.36 | 98 | 311 | 1556 | −0.11 | 108.8 | 67 | 225 | 795 | 0.03 | 0.13 |
| Coordinates in Base Pairs | Offspring-Sex | n° Hetero 1 Dams | A × AB 2 | B × AB | Frequency M/(F) | TRD Parameters | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA/A 3 | AB | B | A | AB | BB/B | |||||
| 4,134,094 | - | 391 | 111 | 46 | 88 | 209 | 150 | 623 | 0.15 (0.18) | αd = −0.12 (BF = 1013.3; ≤0.001 4) |
| 40,239,653 | - | 42,334 | 37,552 | 23,430 | 11,719 | 7816 | 15,685 | 22,229 | 0.64 (0.62) | αd = 0.02 (BF = 1020.6; ≤0.01) |
| 85,463,982 | - | 13,963 | 294 | 241 | 98 | 3401 | 9983 | 14,552 | 0.03 (0.04) | αd = −0.02 (BF = 109.2; ≤0.01) |
| 2,230,488 | M | 640 | 854 | 0 | 668 | 58 | 0 | 39 | 0.94 | αd♂ = 0.06 (BF = 104.14; ≤0.01) |
| F | 477 | 632 | 628 | 0 | 0 | 20 | 18 | 0.93 | αd♀ = 0 (BF = 0.06; >5) | |
| 3,873,466 | M | 81 | 88 | 0 | 56 | 42 | 0 | 20 | 0.52 | αd♂ = 0.13 (BF = 107.2; ≤1) |
| F | 48 | 35 | 25 | 0 | 0 | 19 | 32 | 0.49 | αd♀ = −0.01 (BF = 0.12; >5) | |
| 5,451,082 | M | 8389 | 10,984 | 0 | 10,371 | 1785 | 0 | 1673 | 0.86 | αd♂ = 0.01 (BF = 234.4; ≤0.01) |
| F | 31,944 | 25,802 | 25,820 | 0 | 0 | 3570 | 3826 | 0.85 | αd♀ = 0 (BF = 0.01; >5) | |
| 22,030,046 | M | 1364 | 846 | 0 | 710 | 931 | 0 | 795 | 0.46 | αd♂ = 0.04 (BF = 103.2; ≤1) |
| F | 1067 | 603 | 591 | 0 | 0 | 713 | 672 | 0.43 | αd♀ = 0.01 (BF = 0.04; >5) | |
| 65,136,071 | M | 12,977 | 5812 | 0 | 6482 | 13,607 | 0 | 14,685 | 0.34 | αd♂ = −0.02 (BF = 1014.3; ≤0.001) |
| F | 49,960 | 13,780 | 14,055 | 0 | 0 | 32,898 | 33,032 | 0.37 | αd♀ = 0 (BF = 0.01; >5) | |
| 106,467,781 | M | 1627 | 425 | 0 | 513 | 1658 | 0 | 1943 | 0.21 | αd♂ = −0.04 (BF = 104.93; ≤1) |
| F | 1128 | 305 | 309 | 0 | 0 | 1234 | 1265 | 0.21 | αd♀ = −0.01 (BF = 0.03; >5) | |
| 143,832,372 | M | 13,163 | 7425 | 0 | 7888 | 12,371 | 0 | 13,080 | 0.38 | αd♂ = −0.01 (BF = 104.78; ≤0.01) |
| F | 52,914 | 19,253 | 18,869 | 0 | 0 | 30,256 | 30,222 | 0.44 | αd♀ = 0.0 (BF = 0.01; >5) | |
| 67,099,097 | M | 785 | 1192 | 0 | 1146 | 53 | 0 | 41 | 0.88 | αd♂ = 0.01 (BF = 0.05; >5) |
| F | 504 | 936 | 586 | 0 | 0 | 28 | 37 | 0.91 | αd♀ = 0.11 (BF = 1014.5; ≤0.01) | |
| 125,033,467 | M | 767 | 1116 | 0 | 1082 | 56 | 0 | 41 | 0.95 | αd♂ = 0.01 (BF = 0.04; >5) |
| F | 613 | 880 | 708 | 0 | 0 | 45 | 40 | 0.95 | αd♀ = 0.05 (BF = 363.1; ≤0.1) | |
| Coordinates in Base Pairs 1 | n° Hetero 2 Dams | Dam-TRD | BF | A × AB 3 | B × AB | Frequency % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA/A 4 | AB | B | A | AB | BB/B | |||||
| 134,602,363:135,936,247 4 | 10 | −0.49 | 1019.80 | 0 | 0 | 0 | 0 | 0 | 72 | 0.004 |
| 134,841,753:136,063,445 | 11 | −0.49 | 1019.80 | 0 | 0 | 0 | 0 | 0 | 72 | 0.004 |
| 134,624,675:135,970,186 | 10 | −0.49 | 1019.51 | 0 | 0 | 0 | 0 | 0 | 71 | 0.004 |
| 119,781,376:121,600,055 | 11 | −0.44 | 1019.13 | 0 | 0 | 0 | 1 | 4 | 91 | 0.007 |
| 123,894,981:125,905,926 | 101 | −0.22 | 1018.12 | 0 | 0 | 0 | 46 | 72 | 310 | 0.109 |
| 053,138,058:058,932,871 | 18 | −0.38 | 1016.11 | 0 | 0 | 0 | 3 | 10 | 103 | 0.021 |
| 056,821,723:059,606,508 | 12 | −0.47 | 1013.65 | 0 | 0 | 0 | 0 | 1 | 56 | 0.005 |
| 056,846,129:059,878,165 | 12 | −0.47 | 1013.65 | 0 | 0 | 0 | 0 | 1 | 56 | 0.005 |
| 129,911,571:132,268,728 | 15 | −0.44 | 1013.11 | 0 | 0 | 0 | 0 | 3 | 62 | 0.007 |
| 130,070,306:132,334,180 | 15 | −0.44 | 1013.11 | 0 | 0 | 0 | 0 | 3 | 62 | 0.007 |
| 130,112,536:132,620,448 | 15 | −0.44 | 1013.11 | 0 | 0 | 0 | 0 | 3 | 62 | 0.007 |
| 055,893,493:059,564,066 | 44 | −0.24 | 1012.55 | 0 | 0 | 0 | 31 | 40 | 199 | 0.058 |
| 129,070,088:131,766,182 | 12 | −0.45 | 1011.64 | 0 | 0 | 0 | 1 | 1 | 53 | 0.006 |
| 129,145,800:131,806,987 | 12 | −0.45 | 1011.64 | 0 | 0 | 0 | 1 | 1 | 53 | 0.006 |
| 129,193,110:131,960,783 | 12 | −0.45 | 1011.64 | 0 | 0 | 0 | 1 | 1 | 53 | 0.006 |
| 129,718,409:132,109,716 | 12 | −0.45 | 1011.64 | 0 | 0 | 0 | 1 | 1 | 53 | 0.006 |
| 129,887,133:132,211,189 | 12 | −0.45 | 1011.64 | 0 | 0 | 0 | 1 | 1 | 53 | 0.006 |
| 023,761,259:026,607,222 | 30 | −0.29 | 1011.07 | 0 | 0 | 0 | 5 | 25 | 118 | 0.025 |
| 056,962,509:060,125,574 | 13 | −0.40 | 1010.59 | 0 | 0 | 0 | 2 | 4 | 62 | 0.008 |
| 100,533,235:105,003,475 | 14 | −0.38 | 1010.47 | 0 | 0 | 0 | 5 | 3 | 67 | 0.011 |
| Coordinates in Base Pairs (n. SNPs 1) | n° Hetero 2 Dams | AA × AB 3 | BB × AB | Frequency % | TRD Parameters | ||
|---|---|---|---|---|---|---|---|
| AA 4 | AB | AB | BB | ||||
| 307,557: 854,184 (10) | 247.00 | 2 | 8 | 300 | 281 | 0.40 | αg = −0.34 (BF = 103.35); δg = 0.21; (BF = 103.17) |
| 14,897,141:16,123,681 (10) | 452.00 | 1 | 8 | 559 | 579 | 0.60 | αg = −0.47 (BF = 1012.96); δg = 0.21; (BF = 106.64) |
| 23,895,384:27,387,980 (20) | 541.00 | 1 | 7 | 674 | 634 | 0.90 | αg = −0.40 (BF = 1012.08); δg = 0.24; (BF = 1010.46) |
| 30,116,443:30,459,667 (4) | 574.00 | 0 | 6 | 683 | 689 | 0.90 | αg = −0.51 (BF = 1019.6); δg = 0.25; (BF = 1011.37) |
| 48,750,092:52,808,614 (20) | 592.00 | 0 | 5 | 647 | 669 | 1.10 | αg = −0.50 (BF = 1017.26); δg = 0.23; (BF = 108.99) |
| 59,564,066:61,635,072 (20) | 525.00 | 0 | 8 | 550 | 549 | 1.80 | αg = −0.56 (BF = 1019.61); δg = 0.28; (BF = 1011.44) |
| 109,041,641:110,425,524 (20) | 438.00 | 1 | 8 | 513 | 539 | 1.20 | αg = −0.48 (BF = 1012.4); δg = 0.21; (BF = 105.82) |
| 127,431,846:129,887,133 (20) | 406.00 | 2 | 9 | 573 | 551 | 0.80 | αg = −0.37 (BF = 108.37); δg = 0.21; (BF = 106.78) |
| p-Value | False Discovery Rate | Genes | |
|---|---|---|---|
| Canonical Metabolic Pathway | |||
| Citrulline Degradation | 0.0072 | 0.603 | OTC |
| Spermine Biosynthesis | 0.0145 | 0.603 | SMS |
| PRPP Biosynthesis I | 0.0282 | 0.603 | PRPS1 |
| Arginine Biosynthesis IV | 0.0427 | 0.603 | OTC |
| Proline Biosynthesis II (from Arginine) | 0.0427 | 0.603 | OTC |
| Urea Cycle | 0.0427 | 0.603 | OTC |
| Pentose Phosphate Pathway (Non-oxidative Branch) | 0.0427 | 0.603 | TKTL1 |
| Glycine Cleavage Complex | 0.0427 | 0.603 | GCSH |
| Phosphatidylcholine Biosynthesis I | 0.0490 | 0.603 | PCYT1B |
| Acetyl-CoA Biosynthesis I (Pyruvate Dehydrogenase Complex) | 0.0490 | 0.603 | PDHA1 |
| Diseases and Functions | |||
| X-linked hereditary disease | 3.21 × 10−47 | 1.6 × 10−44 | ADGRG2, ALG13, AMELX, AMER1, ANOS1, AP1S2, ARHGEF9, ARMCX5GPRASP2/GPRASP2, ARR3, BTK, CA5B, CDKL5, CNKSR2, COL4A6, FANCB, FGF13, FLNA, FRMD7, GLA, GPR143, HNRNPH2, IGSF1, KDM6A, KIF4A, MBTPS2, MECP2, MID2, mir221, NDUFA1, OPN1LW, OTC, PCDH19, PDHA1, PDK3, PHEX, PHKA2, PIGA, PIH1D3, PLP1, POLA1, PRPS1, RNF113A, RPS6KA3, RS1, SERPINA7, SMPX, SMS, SRPX2, TIMM8A, TSPAN7, UBE2A, ZC4H2, ZNF674 |
| X-linked mental retardation | 1.47 × 10−15 | 3.7 × 10−13 | AP1S2, CNKSR2, HNRNPH2, KDM6A, KIF4A, MECP2, MID2, PDHA1, RNF113A, RPS6KA3, SMS, SRPX2, TSPAN7, UBE2A, ZC4H2, ZNF674 |
| X-linked hearing loss | 1.29 × 10−8 | 2.2 × 10−6 | ARMCX5GPRASP2/GPRASP2, COL4A6, PRPS1, SMPX |
| Syndromic X-linked mental retardation | 1.97 × 10−8 | 2.5 × 10−6 | AP1S2, CNKSR2, HNRNPH2, KDM6A, MECP2, SMS, SRPX2, UBE2A, ZC4H2 |
| Familial mental retardation | 4.28 × 10−8 | 4.3 × 10−6 | AP1S2, CNKSR2, FLNA, HNRNPH2, KDM6A, KIF4A, MECP2, MID2, PDHA1, RNF113A, RPS6KA3, SMS, SRPX2, TSPAN7, UBE2A, ZC4H2, ZNF674 |
| X-linked nonsyndromic sensorineural hearing loss | 1.46 × 10−6 | 1.2 × 10−4 | COL4A6, PRPS1, SMPX |
| Autism or intellectual disability | 1.95 × 10−6 | 1.4 × 10−4 | AP1S2, CDKL5, CNKSR2, FLNA, HNRNPH2, KDM6A, KIF4A, MECP2, MID2, PDHA1, RNF113A, RPS6KA3, SMS, SRPX2, TSPAN7, UBE2A, ZC4H2, ZNF674 |
| Familial syndromic intellectual disability | 2.57 × 10−6 | 1.6 × 10−4 | AP1S2, CNKSR2, FLNA, HNRNPH2, KDM6A, MECP2, SMS, SRPX2, UBE2A, ZC4H2 |
| Childhood onset epilepsy syndrome | 3.63 × 10−6 | 2.0 × 10−4 | ARHGEF9, CDKL5, SRPX2 |
| Cognitive impairment | 3.49 × 10−5 | 1.7 × 10−3 | AP1S2, ARHGEF9, CA5B, CNKSR2, FLNA, HNRNPH2, KDM6A, KIF4A, MECP2, MID2, PDHA1, RNF113A, RPS6KA3, SMS, SRPX2, TSPAN7, UBE2A, VEGFD, ZC4H2, ZNF674 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Id-Lahoucine, S.; Casellas, J.; Fonseca, P.A.S.; Suárez-Vega, A.; Schenkel, F.S.; Cánovas, A. Deviations from Mendelian Inheritance on Bovine X-Chromosome Revealing Recombination, Sex-of-Offspring Effects and Fertility-Related Candidate Genes. Genes 2022, 13, 2322. https://doi.org/10.3390/genes13122322
Id-Lahoucine S, Casellas J, Fonseca PAS, Suárez-Vega A, Schenkel FS, Cánovas A. Deviations from Mendelian Inheritance on Bovine X-Chromosome Revealing Recombination, Sex-of-Offspring Effects and Fertility-Related Candidate Genes. Genes. 2022; 13(12):2322. https://doi.org/10.3390/genes13122322
Chicago/Turabian StyleId-Lahoucine, Samir, Joaquim Casellas, Pablo A. S. Fonseca, Aroa Suárez-Vega, Flavio S. Schenkel, and Angela Cánovas. 2022. "Deviations from Mendelian Inheritance on Bovine X-Chromosome Revealing Recombination, Sex-of-Offspring Effects and Fertility-Related Candidate Genes" Genes 13, no. 12: 2322. https://doi.org/10.3390/genes13122322