
Folic Acid and Vitamin B12 Prevent Deleterious Effects of Rotenone on Object Novelty Recognition Memory and Kynu Expression in an Animal Model of Parkinson’s Disease

 , , , ,
, , , ,
Abstract
:
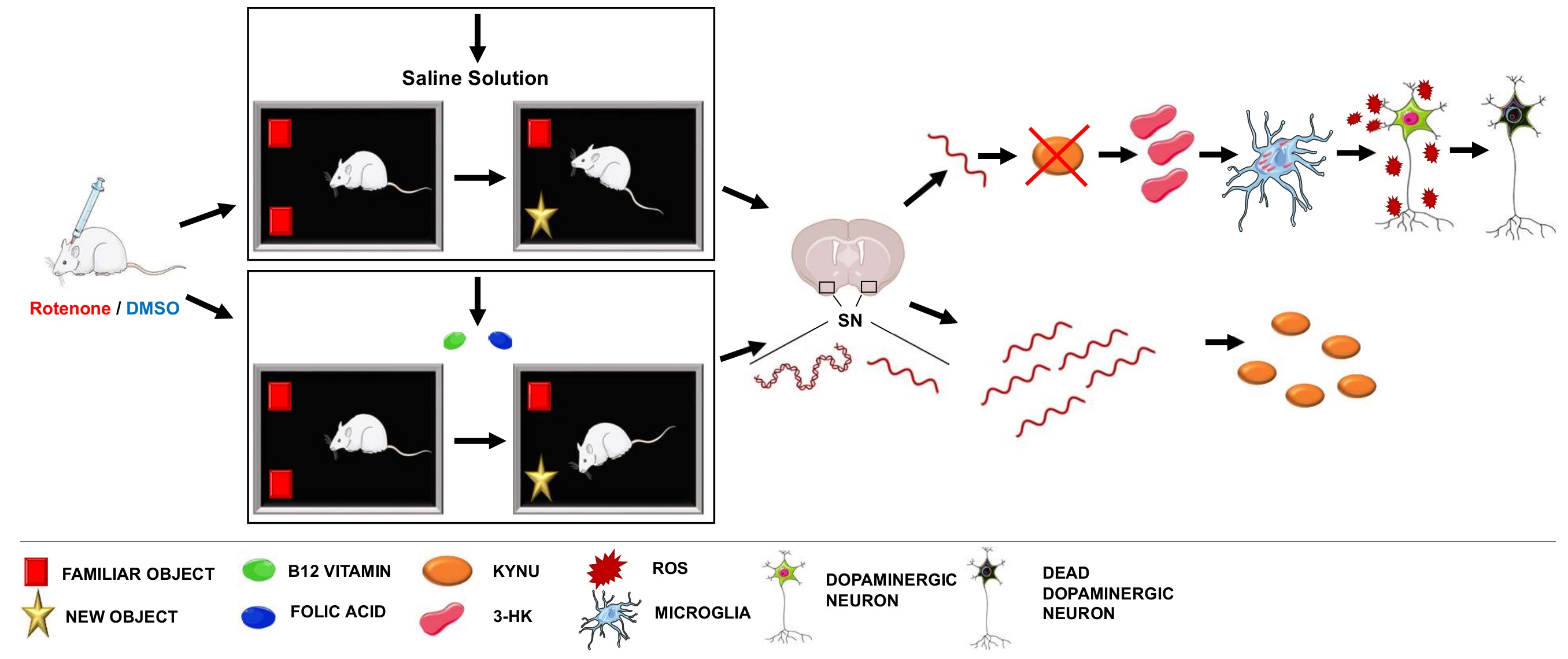{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
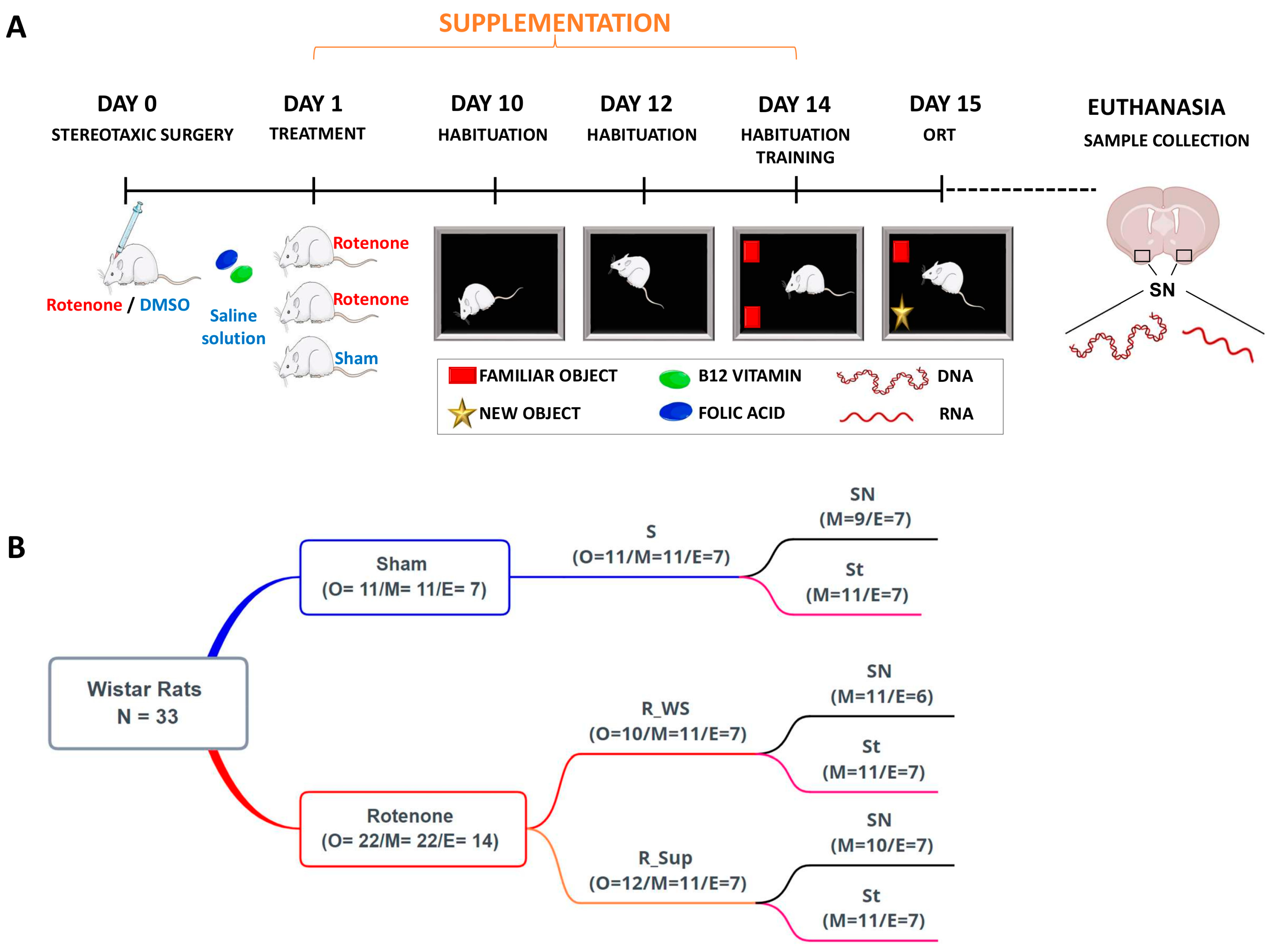
2.1. Animals
2.2. Experimental Design
2.3. Stereotaxic Surgery
2.4. Supplementation Procedure
2.5. Object Recognition Task Test (ORT)
2.6. Gene Regulation and Expression Analysis
2.6.1. DNA Methylation
2.6.2. Gene Expression
2.7. Statistical Analysis
3. Results
3.1. Object Recognition Task Test (ORT)
3.2. Kynu Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells Us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Duda, J.E.; Giasson, B.I.; Mabon, M.E.; Lee, V.M.Y.; Trojanowski, J.Q. Novel Antibodies to Synuclein Show Abundant Striatal Pathology in Lewy Body Diseases. Ann. Neurol. 2002, 52, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A. Parkinson Disease. Contin. Lifelong Learn. Neurol. 2019, 25, 896–918. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Mehan, S.; Singh, S. Understanding Multifactorial Architecture of Parkinson’s Disease: Pathophysiology to Management. Neurol. Sci. 2019, 40, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.V.; Halliday, G. Missing Pieces in the Parkinson’s Disease Puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Ilkiw, J.; Lima, M.S. Perspectives for the Association between Olfactory Disturbances and Depression in Parkinson’s Disease. Neural Regen. Res. 2019, 14, 591. [Google Scholar] [CrossRef]
- Lima, M.M.S. Sleep Disturbances in Parkinson’s Disease: The Contribution of Dopamine in REM Sleep Regulation. Sleep Med. Rev. 2013, 17, 367–375. [Google Scholar] [CrossRef]
- Targa, A.D.S.; Rodrigues, L.S.; Noseda, A.C.D.; Aurich, M.F.; Andersen, M.L.; Tufik, S.; Da Cunha, C.; Lima, M.M.S. Unraveling a New Circuitry for Sleep Regulation in Parkinson’s Disease. Neuropharmacology 2016, 108, 161–171. [Google Scholar] [CrossRef]
- Santos Lima, G.Z.; Targa, A.D.S.; Freitas Cavalcante, S.; Rodrigues, L.S.; Fontenele-Araújo, J.; Torterolo, P.; Andersen, M.L.; Lima, M.M.S. Disruption of Neocortical Synchronisation during Slow-wave Sleep in the Rotenone Model of Parkinson’s Disease. J. Sleep Res. 2021, 30, 1–11. [Google Scholar] [CrossRef]
- Klein, H.; De Jager, P.L. Uncovering the Role of the Methylome in Dementia and Neurodegeneration. Trends Mol. Med. 2016, 22, 687–700. [Google Scholar] [CrossRef]
- Renani, P.G.; Taheri, F.; Rostami, D.; Farahani, N.; Abdolkarimi, H.; Abdollahi, E.; Taghizadeh, E.; Gheibi Hayat, S.M. Involvement of Aberrant Regulation of Epigenetic Mechanisms in the Pathogenesis of Parkinson’s Disease and Epigenetic-based Therapies. J. Cell. Physiol. 2019, 234, 19307–19319. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Morales, E.; Meier, K.; Sandoval-Carrillo, A.; Salas-Pacheco, J.; Vázquez-Cárdenas, P.; Arias-Carrión, O. Implications of DNA Methylation in Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Freeman, D.M.; Lou, D.; Li, Y.; Martos, S.N.; Wang, Z. The Conserved DNMT1-Dependent Methylation Regions in Human Cells Are Vulnerable to Neurotoxicant Rotenone Exposure. Epigenetics and Chromatin 2020, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wüllner, U.; Kaut, O.; DeBoni, L.; Piston, D.; Schmitt, I. DNA Methylation in Parkinson’s Disease. J. Neurochem. 2016, 139, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgone, G.; Currò, M.; Ferlazzo, N.; Parisi, G.; Parnetti, L.; Belcastro, V.; Tambasco, N.; Rossi, A.; Pisani, F.; Calabresi, P.; et al. Coenzyme Q10, Hyperhomocysteinemia and MTHFR C677T Polymorphism in Levodopa-Treated Parkinson’s Disease Patients. NeuroMolecular Med. 2012, 14, 84–90. [Google Scholar] [CrossRef]
- Obeid, R.; Schadt, A.; Dillmann, U.; Kostopoulos, P.; Fassbender, K.; Herrmann, W. Methylation Status and Neurodegenerative Markers in Parkinson Disease. Clin. Chem. 2009, 55, 1852–1860. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Ladenheim, B.; Cutler, R.G.; Kruman, I.I.; Cadet, J.L.; Mattson, M.P. Dietary Folate Deficiency and Elevated Homocysteine Levels Endanger Dopaminergic Neurons in Models of Parkinson’s Disease. J. Neurochem. 2002, 80, 101–110. [Google Scholar] [CrossRef]
- Qiu, F.; Wu, Y.; Cao, H.; Liu, B.; Du, M.; Jiang, H.; Li, S. Changes of Peripheral Nerve Function and Vitamin B12 Level in People With Parkinson’s Disease. Front. Neurol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of Folic Acid and Vitamin B12, Alone and in Combination on Cognitive Function and Inflammatory Factors in the Elderly with Mild Cognitive Impairment: A Single-Blind Experimental Design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef]
- Hinterberger, M.; Fischer, P. Folate and Alzheimer: When Time Matters. J. Neural Transm. 2013, 120, 211–224. [Google Scholar] [CrossRef]
- Percário, S.; Da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; De Nazaré Araújo Moreira, T.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxid. Med. Cell. Longev. 2020, 2020, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.K.; Jadavji, N.M. The Role of One-Carbon Metabolism and Homocysteine in Parkinson’s Disease Onset, Pathology and Mechanisms. Nutr. Res. Rev. 2019, 32, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Jardine, J.; Garrido - Maraver, J.; Loh, S.; Martins, L.M. Folinic Acid Is Neuroprotective in a Fly Model of Parkinson’s Disease Associated with Pink1 Mutations. Matters 2017, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Srivastav, S.; Singh, S.K.; Yadav, A.K.; Srikrishna, S. Folic Acid Supplementation Ameliorates Oxidative Stress, Metabolic Functions and Developmental Anomalies in a Novel Fly Model of Parkinson’s Disease. Neurochem. Res. 2015, 40, 1350–1359. [Google Scholar] [CrossRef]
- McCleery, J.; Abraham, R.P.; Denton, D.A.; Rutjes, A.W.; Chong, L.-Y.; Al-Assaf, A.S.; Griffith, D.J.; Rafeeq, S.; Yaman, H.; Malik, M.A.; et al. Vitamin and Mineral Supplementation for Preventing Dementia or Delaying Cognitive Decline in People with Mild Cognitive Impairment. Cochrane Database Syst. Rev. 2018, 63, 713. [Google Scholar] [CrossRef]
- Araújo, J.R.; Martel, F.; Borges, N.; Araújo, J.M.; Keating, E. Folates and Aging: Role in Mild Cognitive Impairment, Dementia and Depression. Ageing Res. Rev. 2015, 22, 9–19. [Google Scholar] [CrossRef]
- Ajibawo-Aganbi, U.; Saleem, S.; Khan, S.Z.A.; Veliginti, S.; Bastidas, M.V.P.; Lungba, R.M.; Cancarevic, I. Can Nutritional Adequacy Help Evade Neurodegeneration in Older Age? A Review. Cureus 2020, 12, e10921. [Google Scholar] [CrossRef]
- McCarter, S.J.; Stang, C.; Turcano, P.; Mielke, M.M.; Ali, F.; Bower, J.H.; Savica, R. Higher Vitamin B12 Level at Parkinson’s Disease Diagnosis Is Associated with Lower Risk of Future Dementia. Park. Relat. Disord. 2020, 73, 19–22. [Google Scholar] [CrossRef]
- Walker, J.G.; Batterham, P.J.; Mackinnon, A.J.; Jorm, A.F.; Hickie, I.; Fenech, M.; Kljakovic, M.; Crisp, D.; Christensen, H. Oral Folic Acid and Vitamin B-12 Supplementation to Prevent Cognitive Decline in Community-Dwelling Older Adults with Depressive Symptoms—the Beyond Ageing Project: A Randomized Controlled Trial. Am. J. Clin. Nutr. 2012, 95, 194–203. [Google Scholar] [CrossRef] [Green Version]
- McCarter, S.J.; Teigen, L.M.; McCarter, A.R.; Benarroch, E.E.; St. Louis, E.K.; Savica, R. Low Vitamin B12 and Parkinson Disease: Potential Link to Reduced Cholinergic Transmission and Severity of Disease. Mayo Clin. Proc. 2019, 94, 757–762. [Google Scholar] [CrossRef]
- Müller, T.; Möhr, J.D. Long-Term Management of Parkinson’s Disease Using Levodopa Combinations. Expert Opin. Pharmacother. 2018, 19, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Feng, H.; Peng, S.; Xiao, J.; Zhang, J. Association of Plasma Homocysteine, Vitamin B12 and Folate Levels with Cognitive Function in Parkinson’s Disease: A Meta-Analysis. Neurosci. Lett. 2017, 636, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwaluw, N.L.; Dhonukshe-Rutten, R.A.M.; van Wijngaarden, J.P.; Brouwer-Brolsma, E.M.; van de Rest, O.; In ’t Veld, P.H.; Enneman, A.W.; van Dijk, S.C.; Ham, A.C.; Swart, K.M.A.; et al. Results of 2-Year Vitamin B Treatment on Cognitive Performance: Secondary Data from an RCT. Neurology 2014, 83, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-M.; Ye, J.-X.; Mu, J.-S.; Cui, X.-P. Efficacy of Vitamin B Supplementation on Cognition in Elderly Patients With Cognitive-Related Diseases. J. Geriatr. Psychiatry Neurol. 2017, 30, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Malouf, R.; Grimley Evans, J. Folic Acid with or without Vitamin B12 for the Prevention and Treatment of Healthy Elderly and Demented People. Cochrane Database Syst. Rev. 2008, 4, 1–82. [Google Scholar] [CrossRef]
- Craenen, K.; Verslegers, M.; Baatout, S.; Abderrafi Benotmane, M. An Appraisal of Folates as Key Factors in Cognition and Ageing-Related Diseases. Crit. Rev. Food Sci. Nutr. 2020, 60, 722–739. [Google Scholar] [CrossRef]
- Lima, M.M.S.; Martins, E.F.; Delattre, A.M.; Proenca, M.B.; Mori, M.A.; Carabelli, B.; Ferraz, A.C. Motor and Non-Motor Features of Parkinson’s Disease – A Review of Clinical and Experimental Studies. CNS Neurol. Disord. - Drug Targets 2012, 11, 439–449. [Google Scholar] [CrossRef]
- Noseda, A.C.D.; Targa, A.D.S.; Rodrigues, L.S.; Aurich, M.F.; Lima, M.M.S. REM Sleep Deprivation Promotes a Dopaminergic Influence in the Striatal MT2 Anxiolytic-like Effects. Sleep Sci. 2016, 9, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Noseda, A.C.D.; Rodrigues, L.S.; Targa, A.D.S.; Aurich, M.F.; Vital, M.A.B.F.; Da Cunha, C.; Lima, M.M.S. Putative Role of Monoamines in the Antidepressant-like Mechanism Induced by Striatal MT2 Blockade. Behav. Brain Res. 2014, 275, 136–145. [Google Scholar] [CrossRef]
- Rodrigues, L.S.; Noseda, A.C.D.; Targa, A.D.S.; Aurich, M.F.; Lima, M.M.S. Olfaction in Female Wistar Rats Is Influenced by Dopaminergic Periglomerular Neurons after Nigral and Bulbar Lesions. Behav. Pharmacol. 2019, 30, 343–350. [Google Scholar] [CrossRef]
- Targa, A.D.S.; Noseda, A.C.D.; Rodrigues, L.S.; Aurich, M.F.; Lima, M.M.S. REM Sleep Deprivation and Dopaminergic D2 Receptors Modulation Increase Recognition Memory in an Animal Model of Parkinson’s Disease. Behav. Brain Res. 2018, 339, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Mándi, Y.; Vécsei, L. The Kynurenine System and Immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent Advances and New Questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, D.; Iyer, M.; Narayanasamy, A.; Siva, K.; Vellingiri, B. Kynurenine Pathway in Parkinson’s Disease—An Update. eNeurologicalSci 2020, 21, 100270. [Google Scholar] [CrossRef]
- Potter, M.C.; Elmer, G.I.; Bergeron, R.; Albuquerque, E.X.; Guidetti, P.; Wu, H.-Q.; Schwarcz, R. Reduction of Endogenous Kynurenic Acid Formation Enhances Extracellular Glutamate, Hippocampal Plasticity, and Cognitive Behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [Google Scholar] [CrossRef]
- Kozak, R.; Campbell, B.M.; Strick, C.A.; Horner, W.; Hoffmann, W.E.; Kiss, T.; Chapin, D.S.; McGinnis, D.; Abbott, A.L.; Roberts, B.M.; et al. Reduction of Brain Kynurenic Acid Improves Cognitive Function. J. Neurosci. 2014, 34, 10592–10602. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Stone, T.W. The Kynurenine Pathway and the Brain: Challenges, Controversies and Promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.K.; Fernández-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the Kynurenine Pathway in the Pathogenesis of Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The Involvement of Neuroinflammation and Kynurenine Pathway in Parkinson’s Disease. Parkinsons. Dis. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the Mammalian Brain: When Physiology Meets Pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Maitre, M.; Klein, C.; Patte-Mensah, C.; Mensah-Nyagan, A.-G. Tryptophan Metabolites Modify Brain Aβ Peptide Degradation: A Role in Alzheimer’s Disease? Prog. Neurobiol. 2020, 190, 101800. [Google Scholar] [CrossRef]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the Immune Response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Rassoulpour, A.; Wu, H.-Q.; Ferre, S.; Schwarcz, R. Nanomolar Concentrations of Kynurenic Acid Reduce Extracellular Dopamine Levels in the Striatum. J. Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef]
- Okuno, A.; Fukuwatari, T.; Shibata, K. High Tryptophan Diet Reduces Extracellular Dopamine Release via Kynurenic Acid Production in Rat Striatum. J. Neurochem. 2011, 118, 796–805. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine Pathway Abnormalities in Parkinson’s Disease. Neurology 1992, 42, 1702. [Google Scholar] [CrossRef]
- Heilman, P.L.; Wang, E.W.; Lewis, M.M.; Krzyzanowski, S.; Capan, C.D.; Burmeister, A.R.; Du, G.; Escobar Galvis, M.L.; Brundin, P.; Huang, X.; et al. Tryptophan Metabolites Are Associated With Symptoms and Nigral Pathology in Parkinson’s Disease. Mov. Disord. 2020, 35, 2028–2037. [Google Scholar] [CrossRef]
- Chang, K.H.; Cheng, M.L.; Tang, H.Y.; Huang, C.Y.; Wu, Y.R.; Chen, C.M. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 6319–6328. [Google Scholar] [CrossRef]
- Havelund, J.F.; Andersen, A.D.; Binzer, M.; Blaabjerg, M.; Heegaard, N.H.H.; Stenager, E.; Faergeman, N.J.; Gramsbergen, J.B. Changes in Kynurenine Pathway Metabolism in Parkinson Patients with L-DOPA-Induced Dyskinesia. J. Neurochem. 2017, 142, 756–766. [Google Scholar] [CrossRef]
- Sorgdrager, F.J.H.; Vermeiren, Y.; Van Faassen, M.; van der Ley, C.; Nollen, E.A.A.; Kema, I.P.; De Deyn, P.P. Age- and Disease-Specific Changes of the Kynurenine Pathway in Parkinson’s and Alzheimer’s Disease. J. Neurochem. 2019, 151, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeWitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. 3-Hydroxykynurenine and Other Parkinson’s Disease Biomarkers Discovered by Metabolomic Analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Shi, Y.; Chen, W.; Jia, X.; Asakawa, T. Can Kynurenine Pathway Be Considered as a Next-Generation Therapeutic Target for Parkinson’s Disease? An Update Information. Biosci. Trends 2022, 16, 2022.01352. [Google Scholar] [CrossRef] [PubMed]
- Boros, F.A.; Vécsei, L. Tryptophan 2,3-Dioxygenase, a Novel Therapeutic Target for Parkinson’s Disease. Expert Opin. Ther. Targets 2021, 25, 877–888. [Google Scholar] [CrossRef]
- Bai, J.; Zheng, Y.; Yu, Y. Urinary Kynurenine as a Biomarker for Parkinson’s Disease. Neurol. Sci. 2021, 42, 697–703. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Zengin, G.; Bumbu, A.G.; Andronie-Cioara, F.L.; Nechifor, A.C.; et al. The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 6737. [Google Scholar] [CrossRef]
- Fagotti, J.; Targa, A.D.S.; Rodrigues, L.S.; Noseda, A.C.D.; Dorieux, F.W.C.; Scarante, F.F.; Ilkiw, J.L.; Louzada, F.M.; Chowdhury, N.R.; van der Veen, D.R.; et al. Chronic Sleep Restriction in the Rotenone Parkinson’s Disease Model in Rats Reveals Peripheral Early-Phase Biomarkers. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Harden, J.L.; Lewis, S.M.; Lish, S.R.; Suárez-Fariñas, M.; Gareau, D.; Lentini, T.; Johnson-Huang, L.M.; Krueger, J.G.; Lowes, M.A. The Tryptophan Metabolism Enzyme L-Kynureninase Is a Novel Inflammatory Factor in Psoriasis and Other Inflammatory Diseases. J. Allergy Clin. Immunol. 2016, 137, 1830–1840. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates—The New Coronal Set; Academic press: Cambridge, MA, USA, 2004; p. 209. [Google Scholar] [CrossRef]
- Tamaddonfard, E.; Samadi, F.; Egdami, K. The Effects of Vitamin B12 and Diclofenac and Their Combination on Cold and Mechanical Allodynia in a Neuropathic Pain Model in Rats. Vet. Res. forum an Int. Q. J. 2013, 4, 19–24. [Google Scholar]
- Rasic-Markovic, A.; Rankov-Petrovic, B.; Hrncic, D.; Krstic, D.; Colovic, M.; Macut, D.; Djuric, D.; Stanojlovic, O. The Effect of Subchronic Supplementation with Folic Acid on Homocysteine Induced Seizures. Acta Physiol. Hung. 2015, 102, 151–162. [Google Scholar] [CrossRef]
- Ennaceur, A.; Delacour, J. A New One-Trial Test for Neurobiological Studies of Memory in Rats. 1: Behavioral Data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Deshpande, K.; Stinnett, G.S.; Seasholtz, A.F.; Murphy, G.G. Conversion of Short-Term to Long-Term Memory in the Novel Object Recognition Paradigm. Neurobiol. Learn. Mem. 2013, 105, 174–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.O.; Liu, Y.; Ryan, C.; Joyce, C.E.; Duan, S.; Cao, L.; Martin, A.; Liao, W.; Menter, A.; Bowcock, A.M. A Subset of Methylated CpG Sites Differentiate Psoriatic from Normal Skin - Supplm. J. Invest. Dermatol. 2012, 132, 583–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfving, B.; Müller, H.K.; Oliveras, I.; Østerbøg, T.B.; Rio-Alamos, C.; Sanchez-Gonzalez, A.; Tobeña, A.; Fernandez-Teruel, A.; Aznar, S. Differential Expression of Synaptic Markers Regulated during Neurodevelopment in a Rat Model of Schizophrenia-like Behavior. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2019, 95, 109669. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J.R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- R Development Core Team, R. R: A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2011, 1, 409. [Google Scholar]
- Rhead, B.; Karolchik, D.; Kuhn, R.M.; Hinrichs, A.S.; Zweig, A.S.; Fujita, P.A.; Diekhans, M.; Smith, K.E.; Rosenbloom, K.R.; Raney, B.J.; et al. The UCSC Genome Browser Database: Update 2010. Nucleic Acids Res. 2010, 38, D613–D619. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting Effective MicroRNA Target Sites in Mammalian MRNAs. Elife 2015, 4, 1–38. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A Decade-Long Collection of Experimentally Supported MiRNA–Gene Interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Andrés-León, E.; González Peña, D.; Gómez-López, G.; Pisano, D.G. MiRGate: A Curated Database of Human, Mouse and Rat MiRNA–MRNA Targets. Database 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a Reference Set of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Yuan, J.; Li, H.; Li, M.; Zhao, G.; Bu, D.; Zhu, W.; Wu, W.; Chen, R.; Zhao, Y. NONCODEv4: Exploring the World of Long Non-Coding RNA Genes. Nucleic Acids Res. 2014, 42, D98–D103. [Google Scholar] [CrossRef] [Green Version]
- Christine, C.W.; Auinger, P.; Joslin, A.; Yelpaala, Y.; Green, R. Vitamin B12 and Homocysteine Levels Predict Different Outcomes in Early Parkinson’s Disease. Mov. Disord. 2018, 33, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Christine, C.W.; Auinger, P.; Saleh, N.; Tian, M.; Bottiglieri, T.; Arning, E.; Tran, N.K.; Ueland, P.M.; Green, R. Relationship of Cerebrospinal Fluid Vitamin B12 Status Markers With Parkinson’s Disease Progression. Mov. Disord. 2020, mds.28073. [Google Scholar] [CrossRef]
- Luthra, N.S.; Marcus, A.H.; Hills, N.K.; Christine, C.W. Vitamin B12 Measurements across Neurodegenerative Disorders. J. Clin. Mov. Disord. 2020, 7, 1–6. [Google Scholar] [CrossRef]
- Dietiker, C.; Kim, S.; Zhang, Y.; Christine, C.W. Characterization of Vitamin B12 Supplementation and Correlation with Clinical Outcomes in a Large Longitudinal Study of Early Parkinson’s Disease. J. Mov. Disord. 2019, 12, 91–96. [Google Scholar] [CrossRef]
- Dong, B.; Wu, R. Plasma Homocysteine, Folate and Vitamin B12 Levels in Parkinson’s Disease in China: A Meta-Analysis. Clin. Neurol. Neurosurg. 2020, 188, 105587. [Google Scholar] [CrossRef]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.-O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-Dopa-Induced Motor and Non-Motor Complications in Parkinson’s Disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef]
- Muller, T. Benefit of Folic Acid Supplementation in Parkinsonian Patients Treated with Levodopa. J. Neurol. Neurosurg. Psychiatry 2003, 74, 549. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, G.; Qureshi, A.; Devrajani, B.; Chippa, M.; Syed, S. Is the Deficiency of Vitamin B12 Related to Oxidative Stress and Neurotoxicity in Parkinsons Patients? CNS Neurol. Disord. - Drug Targets 2008, 7, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.R.; Mattson, M.P. Homocysteine Elicits a DNA Damage Response in Neurons That Promotes Apoptosis and Hypersensitivity to Excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.M.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.A.; et al. The Epigenetics of Aging and Neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, H.; Jin, P. Epigenetics-Based Therapeutics for Neurodegenerative Disorders. Curr. Geriatr. Reports 2012, 1, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Boelens Keun, J.T.; Arnoldussen, I.A.; Vriend, C.; van de Rest, O. Dietary Approaches to Improve Efficacy and Control Side Effects of Levodopa Therapy in Parkinson’s Disease: A Systematic Review. Adv. Nutr. 2021, 12, 2265–2287. [Google Scholar] [CrossRef]
- Anamnart, C.; Kitjarak, R. Effects of Vitamin B12, Folate, and Entacapone on Homocysteine Levels in Levodopa-Treated Parkinson’s Disease Patients: A Randomized Controlled Study. J. Clin. Neurosci. 2021, 88, 226–231. [Google Scholar] [CrossRef]
- Fang, C.; Lv, L.; Mao, S.; Dong, H.; Liu, B. Cognition Deficits in Parkinson’s Disease: Mechanisms and Treatment. Parkinsons. Dis. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bäckström, D.; Granåsen, G.; Domellöf, M.E.; Linder, J.; Jakobson Mo, S.; Riklund, K.; Zetterberg, H.; Blennow, K.; Forsgren, L. Early Predictors of Mortality in Parkinsonism and Parkinson Disease. Neurology 2018, 91, e2045–e2056. [Google Scholar] [CrossRef] [Green Version]
- Hobson, P.; Meara, J.; Ishihara-Paul, L. The Estimated Life Expectancy in a Community Cohort of Parkinson’s Disease Patients with and without Dementia, Compared with the UK Population. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-Kynurenine Pathway Enzymes Are Therapeutic Target for Neuropsychiatric Diseases: Focus on Cell Type Differences. Neuropharmacology 2017, 112, 264–274. [Google Scholar] [CrossRef]
- MS Lima, M.; L Andersen, M.; B Reksidler, A.; C Ferraz, A.; ABF Vital, M.; Tufik, S. Paradoxical Sleep Deprivation Modulates Tyrosine Hydroxylase Expression in the Nigrostriatal Pathway and Attenuates Motor Deficits Induced by Dopaminergic Depletion. CNS Neurol. Disord. Targets (Formerly Curr. Drug Targets-CNS Neurol. Disord. 2012, 11, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; Zhang, L.; Khrebtukova, I.; Reinhardt, M.; Wang, C.; Therneau, T.M.; Banck, M.S.; Schroth, G.P.; Beutler, A.S. Diametrically Opposite Methylome-Transcriptome Relationships in High- and Low-CpG Promoter Genes in Postmitotic Neural Rat Tissue. Epigenetics 2012, 7, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, J.; Guan, T.; Yao, H.; Zhang, W.; Guan, Z.; Wang, Y. MiRNAs and Target Genes in the Blood as Biomarkers for the Early Diagnosis of Parkinson’s Disease. BMC Syst. Biol. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Bender, D.A.; McCreanor, G.M. The Preferred Route of Kynurenine Metabolism in the Rat. Biochim. Biophys. Acta - Gen. Subj. 1982, 717, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, K.; Otsuka, C.; Maeda, T.; Yamahara, K.; Kato, K.; Takahashi, K.; Takahashi, K.; Terayama, Y. Impaired Metabolism of Kynurenine and Its Metabolites in CSF of Parkinson’s Disease. Neurosci. Lett. 2020, 714, 134576. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.-T.; Tan, L. The Kynurenine Pathway in Neurodegenerative Diseases: Mechanistic and Therapeutic Considerations. J. Neurol. Sci. 2012, 323, 1–8. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative Stress: A Major Pathogenesis and Potential Therapeutic Target of Antioxidative Agents in Parkinson’s Disease and Alzheimer’s Disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretzschmar, G.C.; Targa, A.D.S.; Soares-Lima, S.C.; dos Santos, P.I.; Rodrigues, L.S.; Macedo, D.A.; Ribeiro Pinto, L.F.; Lima, M.M.S.; Boldt, A.B.W. Folic Acid and Vitamin B12 Prevent Deleterious Effects of Rotenone on Object Novelty Recognition Memory and Kynu Expression in an Animal Model of Parkinson’s Disease. Genes 2022, 13, 2397. https://doi.org/10.3390/genes13122397
Kretzschmar GC, Targa ADS, Soares-Lima SC, dos Santos PI, Rodrigues LS, Macedo DA, Ribeiro Pinto LF, Lima MMS, Boldt ABW. Folic Acid and Vitamin B12 Prevent Deleterious Effects of Rotenone on Object Novelty Recognition Memory and Kynu Expression in an Animal Model of Parkinson’s Disease. Genes. 2022; 13(12):2397. https://doi.org/10.3390/genes13122397
Chicago/Turabian StyleKretzschmar, Gabriela Canalli, Adriano D. S. Targa, Sheila Coelho Soares-Lima, Priscila Ianzen dos Santos, Lais S. Rodrigues, Daniel A. Macedo, Luis Felipe Ribeiro Pinto, Marcelo M. S. Lima, and Angelica Beate Winter Boldt. 2022. "Folic Acid and Vitamin B12 Prevent Deleterious Effects of Rotenone on Object Novelty Recognition Memory and Kynu Expression in an Animal Model of Parkinson’s Disease" Genes 13, no. 12: 2397. https://doi.org/10.3390/genes13122397