
LMNA Mutation in a Family with a Strong History of Sudden Cardiac Death

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
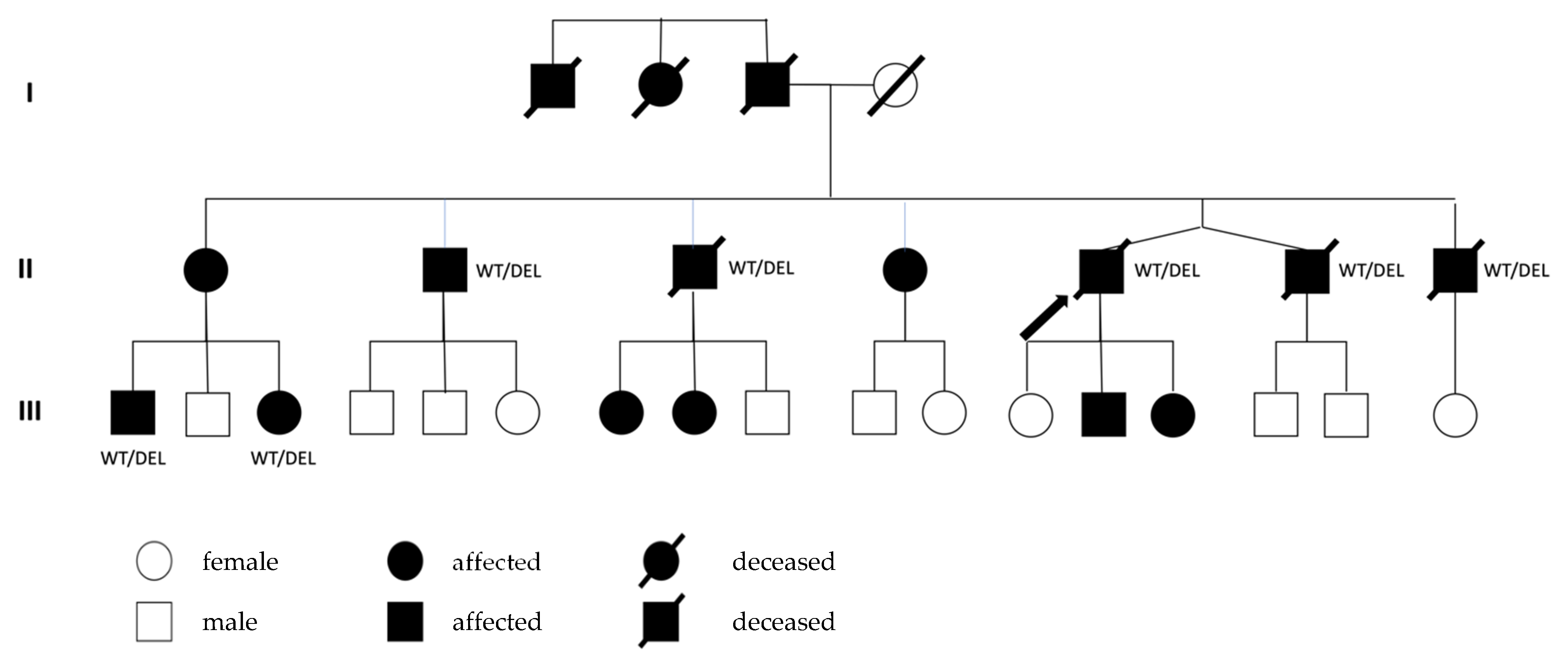
2. Case Presentation
2.1. Clinical Presentation
2.2. Genetic Findings
3. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, M.R.G.; Fain, P.R.; Sinagra, G.; Robinson, M.L.; Robertson, A.D.; Carniel, E.; Di Lenarda, A.; Bohlmeyer, T.J.; Ferguson, D.A.; Brodsky, G.L.; et al. Natural History of Dilated Cardiomyopathy Due to Lamin A/C Gene Mutations. J. Am. Coll. Cardiol. 2003, 41, 771–780. [Google Scholar] [CrossRef] [Green Version]
- van Tintelen, J.P.; Hofstra, R.M.W.; Katerberg, H.; Rossenbacker, T.; Wiesfeld, A.C.P.; du Marchie Sarvaas, G.J.; Wilde, A.A.M.; van Langen, I.M.; Nannenberg, E.A.; van der Kooi, A.J.; et al. High Yield of LMNA Mutations in Patients with Dilated Cardiomyopathy and/or Conduction Disease Referred to Cardiogenetics Outpatient Clinics. Am. Heart J. 2007, 154, 1130–1139. [Google Scholar] [CrossRef]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction-System Disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-Analysis of Clinical Characteristics of 299 Carriers of LMNA Gene Mutations: Do Lamin A/C Mutations Portend a High Risk of Sudden Death? J. Mol. Med. Berl. Ger. 2005, 83, 79–83. [Google Scholar] [CrossRef] [PubMed]
- van Rijsingen, I.A.W.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk Factors for Malignant Ventricular Arrhythmias in Lamin a/c Mutation Carriers a European Cohort Study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Hasselberg, N.E.; Edvardsen, T.; Petri, H.; Berge, K.E.; Leren, T.P.; Bundgaard, H.; Haugaa, K.H. Risk Prediction of Ventricular Arrhythmias and Myocardial Function in Lamin A/C Mutation Positive Subjects. EP Eur. 2014, 16, 563–571. [Google Scholar] [CrossRef]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Bécane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the Gene Encoding Lamin A/C Cause Autosomal Dominant Emery-Dreifuss Muscular Dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Bonne, G.; van der Kooi, A.J.; van Meegen, M.; Baas, F.; Bolhuis, P.A.; de Visser, M.; Schwartz, K. Identification of Mutations in the Gene Encoding Lamins A/C in Autosomal Dominant Limb Girdle Muscular Dystrophy with Atrioventricular Conduction Disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Taylor, M.R.G. Lamin A/C Gene and the Heart: How Genetics May Impact Clinical Care. J. Am. Coll. Cardiol. 2008, 52, 1261–1262. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, C.J. Lamins: Building Blocks or Regulators of Gene Expression? Nat. Rev. Mol. Cell Biol. 2002, 3, 848–858. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic Reorganization of Lamin-Associated Domains in Cardiac Myocytes Is Associated With Differential Gene Expression and DNA Methylation in Human Dilated Cardiomyopathy. Circ. Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef]
- Marian, A.J.; Asatryan, B.; Wehrens, X.H.T. Genetic Basis and Molecular Biology of Cardiac Arrhythmias in Cardiomyopathies. Cardiovasc. Res. 2020, 116, 1600–1619. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin Mutation Induces Conduction Defects through Epigenetic Inhibition of SCN5A in Human Cardiac Laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef] [Green Version]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Richards, A.A.; Zhu, X.; Joglar, J.A.; Yin, H.L.; Garg, V. A Novel Mutation in LAMIN A/C Is Associated with Isolated Early-Onset Atrial Fibrillation and Progressive Atrioventricular Block Followed by Cardiomyopathy and Sudden Cardiac Death. Heart Rhythm 2009, 6, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Peng, D. Case Series: LMNA-Related Dilated Cardiomyopathy Presents with Reginal Wall Akinesis and Transmural Late Gadolinium Enhancement. ESC Heart Fail. 2020, 7, 3179–3183. [Google Scholar] [CrossRef]
- Chen, W.; Huo, J.; Ma, A.; Bai, L.; Liu, P. A Novel Mutation of the LMNA Gene in a Family with Dilated Cardiomyopathy, Conduction System Disease, and Sudden Cardiac Death of Young Females. Mol. Cell. Biochem. 2013, 382, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.; Torretta, S.; Procino, G.; Gerbino, A.; Forleo, C.; Favale, S.; Svelto, M. Role of Nuclear Lamin A/C in Cardiomyocyte Functions. Biol. Cell 2014, 106, 346–358. [Google Scholar] [CrossRef]
- Gerbino, A.; Procino, G.; Svelto, M.; Carmosino, M. Role of Lamin A/C Gene Mutations in the Signaling Defects Leading to Cardiomyopathies. Front. Physiol. 2018, 9, 1356. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; McKenna, W.J. Sudden Death from Genetic and Acquired Cardiomyopathies. Circulation 2012, 125, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacène, E.; Fromes, Y.; Toussaint, M.; Mura, A.-M.; Keller, D.I.; et al. Mouse Model Carrying H222P-Lmna Mutation Develops Muscular Dystrophy and Dilated Cardiomyopathy Similar to Human Striated Muscle Laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 Directly Acts on CTGF/CCN2 Expression to Mediate Myocardial Fibrosis in Cardiomyopathy Caused by Mutations in the Lamin A/C Gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef] [Green Version]
- Captur, G.; Arbustini, E.; Syrris, P.; Radenkovic, D.; O’Brien, B.; Mckenna, W.J.; Moon, J.C. Lamin Mutation Location Predicts Cardiac Phenotype Severity: Combined Analysis of the Published Literature. Open Heart 2018, 5, e000915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyhan-Birsoy, O.; Pugh, T.J.; Bowser, M.J.; Hynes, E.; Frisella, A.L.; Mahanta, L.M.; Lebo, M.S.; Amr, S.S.; Funke, B.H. Next Generation Sequencing-Based Copy Number Analysis Reveals Low Prevalence of Deletions and Duplications in 46 Genes Associated with Genetic Cardiomyopathies. Mol. Genet. Genomic Med. 2016, 4, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Bilinska, Z.T.; Sylvius, N.; Boudreau, E.; Veinot, J.P.; Labib, S.; Bolongo, P.M.; Hamza, A.; Jackson, T.; Ploski, R.; et al. Genetic and Ultrastructural Studies in Dilated Cardiomyopathy Patients: A Large Deletion in the Lamin A/C Gene Is Associated with Cardiomyocyte Nuclear Envelope Disruption. Basic Res. Cardiol. 2010, 105, 365–377. [Google Scholar] [CrossRef] [Green Version]
- van Tintelen, J.P.; Tio, R.A.; Kerstjens-Frederikse, W.S.; van Berlo, J.H.; Boven, L.G.; Suurmeijer, A.J.H.; White, S.J.; den Dunnen, J.T.; te Meerman, G.J.; Vos, Y.J.; et al. Severe Myocardial Fibrosis Caused by a Deletion of the 5′ End of the Lamin A/C Gene. J. Am. Coll. Cardiol. 2007, 49, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Cirino, A.L.; Cuddy, S.; Lakdawala, N.K. Deletion of Entire LMNA Gene as a Cause of Cardiomyopathy. Hear. Case Rep. 2020, 6, 395–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegele, R. LMNA Mutation Position Predicts Organ System Involvement in Laminopathies. Clin. Genet. 2005, 68, 31–34. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C Cardiomyopathy: Young Onset, High Penetrance, and Frequent Need for Heart Transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal Dominant Dilated Cardiomyopathy with Atrioventricular Block: A Lamin A/C Defect-Related Disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Anselme, F.; Moubarak, G.; Savouré, A.; Godin, B.; Borz, B.; Drouin-Garraud, V.; Gay, A. Implantable Cardioverter-Defibrillators in Lamin A/C Mutation Carriers with Cardiac Conduction Disorders. Heart Rhythm 2013, 10, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Glikson, M.; Nielsen, J.C.; Kronborg, M.B.; Michowitz, Y.; Auricchio, A.; Barbash, I.M.; Barrabés, J.A.; Boriani, G.; Braunschweig, F.; Brignole, M.; et al. 2021 ESC Guidelines on Cardiac Pacing and Cardiac Resynchronization Therapy. Eur. Heart J. 2021, 42, 3427–3520. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C Gene Mimic Arrhythmogenic Right Ventricular Cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Forleo, C.; Carmosino, M.; Resta, N.; Rampazzo, A.; Valecce, R.; Sorrentino, S.; Iacoviello, M.; Pisani, F.; Procino, G.; Gerbino, A.; et al. Clinical and Functional Characterization of a Novel Mutation in Lamin a/c Gene in a Multigenerational Family with Arrhythmogenic Cardiac Laminopathy. PLoS ONE 2015, 10, e0121723. [Google Scholar] [CrossRef]
- Holmström, M.; Kivistö, S.; Heliö, T.; Jurkko, R.; Kaartinen, M.; Antila, M.; Reissell, E.; Kuusisto, J.; Kärkkäinen, S.; Peuhkurinen, K.; et al. Late Gadolinium Enhanced Cardiovascular Magnetic Resonance of Lamin A/C Gene Mutation Related Dilated Cardiomyopathy. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson. 2011, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Peretto, G.; Barison, A.; Forleo, C.; Di Resta, C.; Esposito, A.; Aquaro, G.D.; Scardapane, A.; Palmisano, A.; Emdin, M.; Resta, N.; et al. Late Gadolinium Enhancement Role in Arrhythmic Risk Stratification of Patients with LMNA Cardiomyopathy: Results from a Long-Term Follow-up Multicentre Study. EP Eur. 2020, 22, 1864–1872. [Google Scholar] [CrossRef]
- Di Marco, A.; Anguera, I.; Schmitt, M.; Klem, I.; Neilan, T.G.; White, J.A.; Sramko, M.; Masci, P.G.; Barison, A.; Mckenna, P.; et al. Late Gadolinium Enhancement and the Risk for Ventricular Arrhythmias or Sudden Death in Dilated Cardiomyopathy: Systematic Review and Meta-Analysis. JACC Heart Fail. 2017, 5, 28–38. [Google Scholar] [CrossRef]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated Cardiomyopathy and Arrhythmogenic Left Ventricular Cardiomyopathy: A Comprehensive Genotype-Imaging Phenotype Study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keil, L.; Berisha, F.; Knappe, D.; Kubisch, C.; Shoukier, M.; Kirchhof, P.; Fabritz, L.; Hellenbroich, Y.; Woitschach, R.; Magnussen, C. LMNA Mutation in a Family with a Strong History of Sudden Cardiac Death. Genes 2022, 13, 169. https://doi.org/10.3390/genes13020169
Keil L, Berisha F, Knappe D, Kubisch C, Shoukier M, Kirchhof P, Fabritz L, Hellenbroich Y, Woitschach R, Magnussen C. LMNA Mutation in a Family with a Strong History of Sudden Cardiac Death. Genes. 2022; 13(2):169. https://doi.org/10.3390/genes13020169
Chicago/Turabian StyleKeil, Laura, Filip Berisha, Dorit Knappe, Christian Kubisch, Moneef Shoukier, Paulus Kirchhof, Larissa Fabritz, Yorck Hellenbroich, Rixa Woitschach, and Christina Magnussen. 2022. "LMNA Mutation in a Family with a Strong History of Sudden Cardiac Death" Genes 13, no. 2: 169. https://doi.org/10.3390/genes13020169
APA StyleKeil, L., Berisha, F., Knappe, D., Kubisch, C., Shoukier, M., Kirchhof, P., Fabritz, L., Hellenbroich, Y., Woitschach, R., & Magnussen, C. (2022). LMNA Mutation in a Family with a Strong History of Sudden Cardiac Death. Genes, 13(2), 169. https://doi.org/10.3390/genes13020169