
Epigenomic Modifications in Modern and Ancient Genomes

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epigenetic Mechanisms
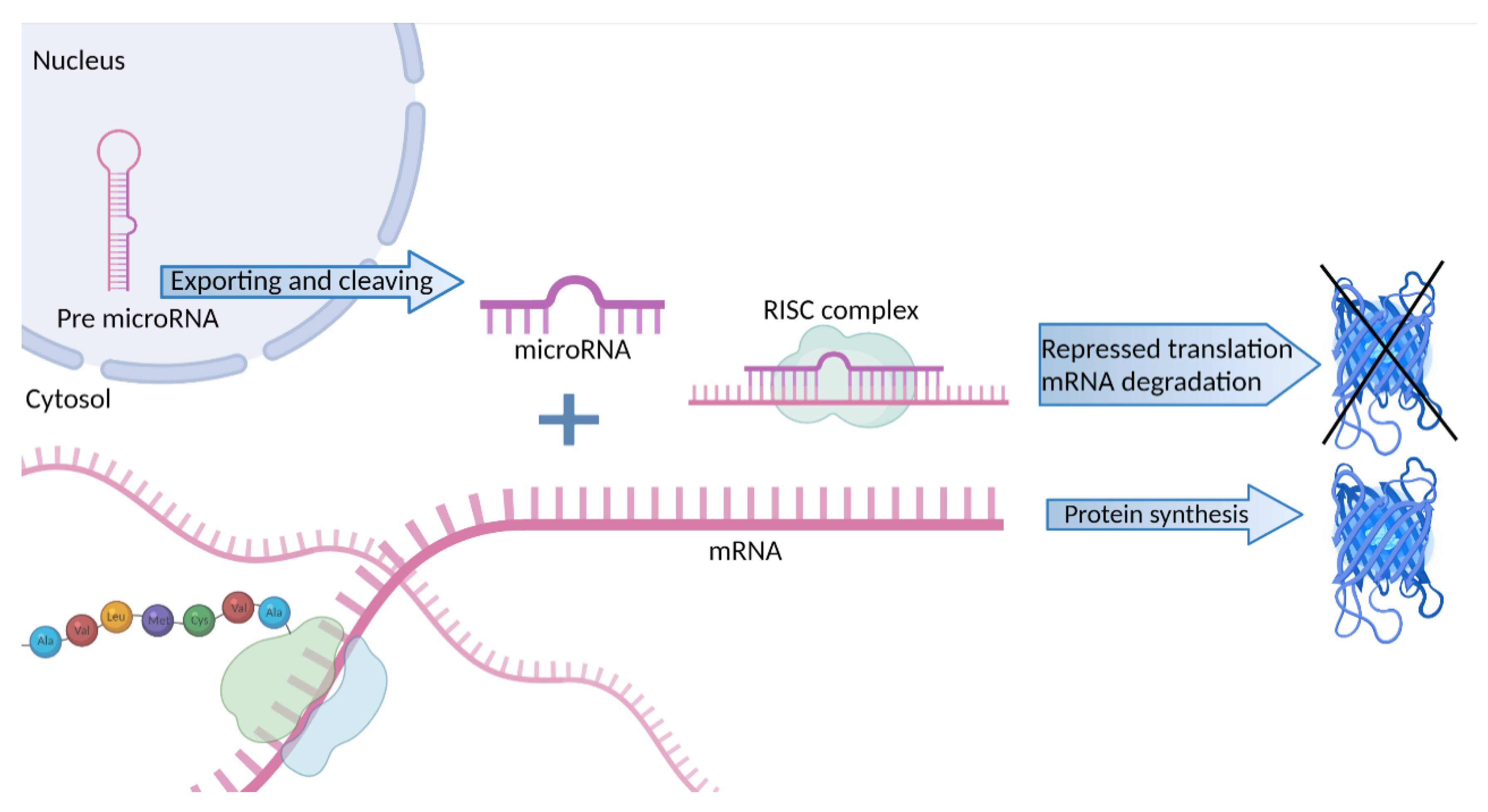
2.1. MicroRNA Silencing Activity
2.2. Histone Modifications
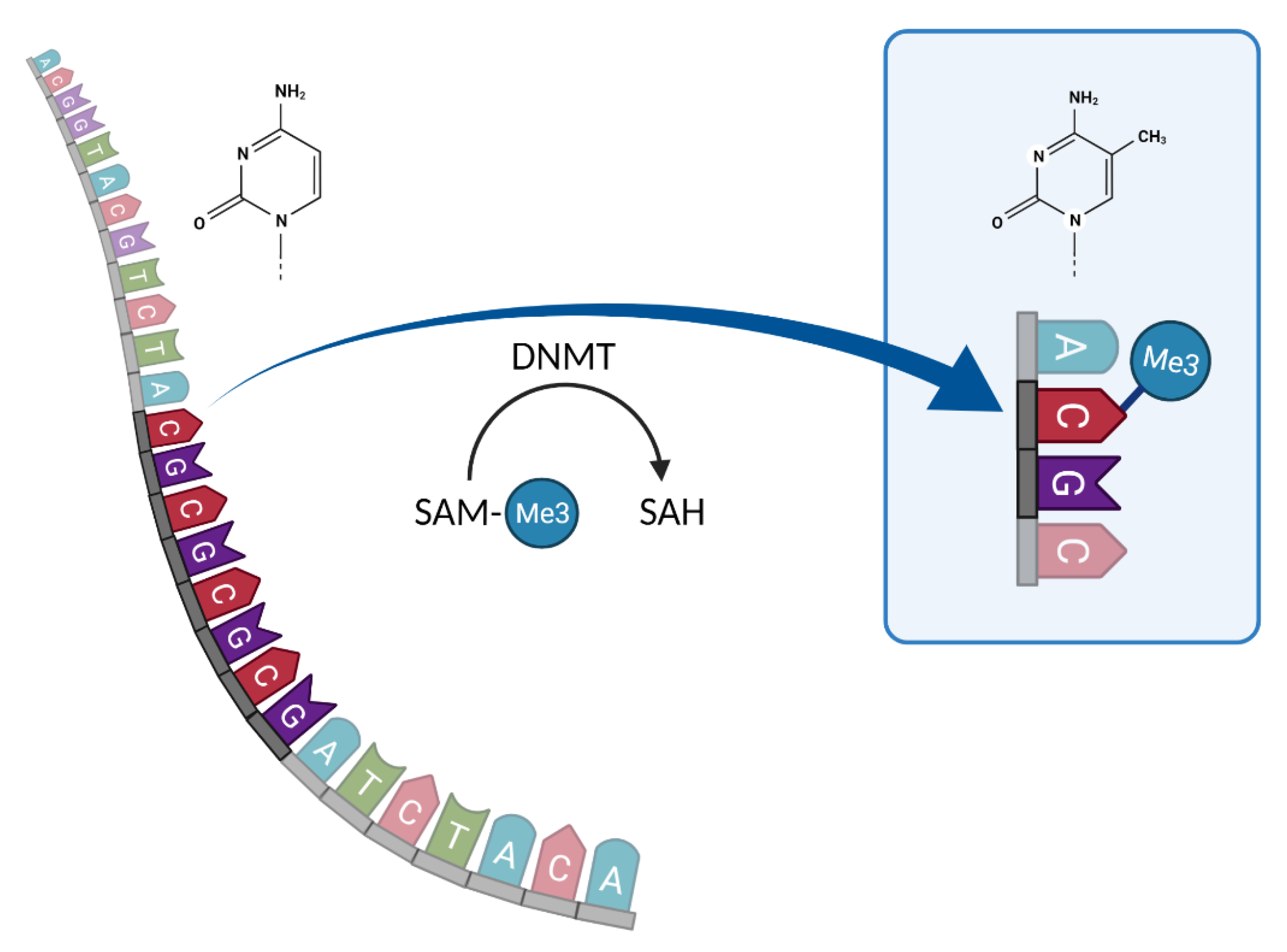
2.3. DNA Methylation
Effects of Environment on Methylation
3. Paleoepigenomics
3.1. Ancient DNA (aDNA) Challenges
3.2. Human Paleoepigenomic Case Studies
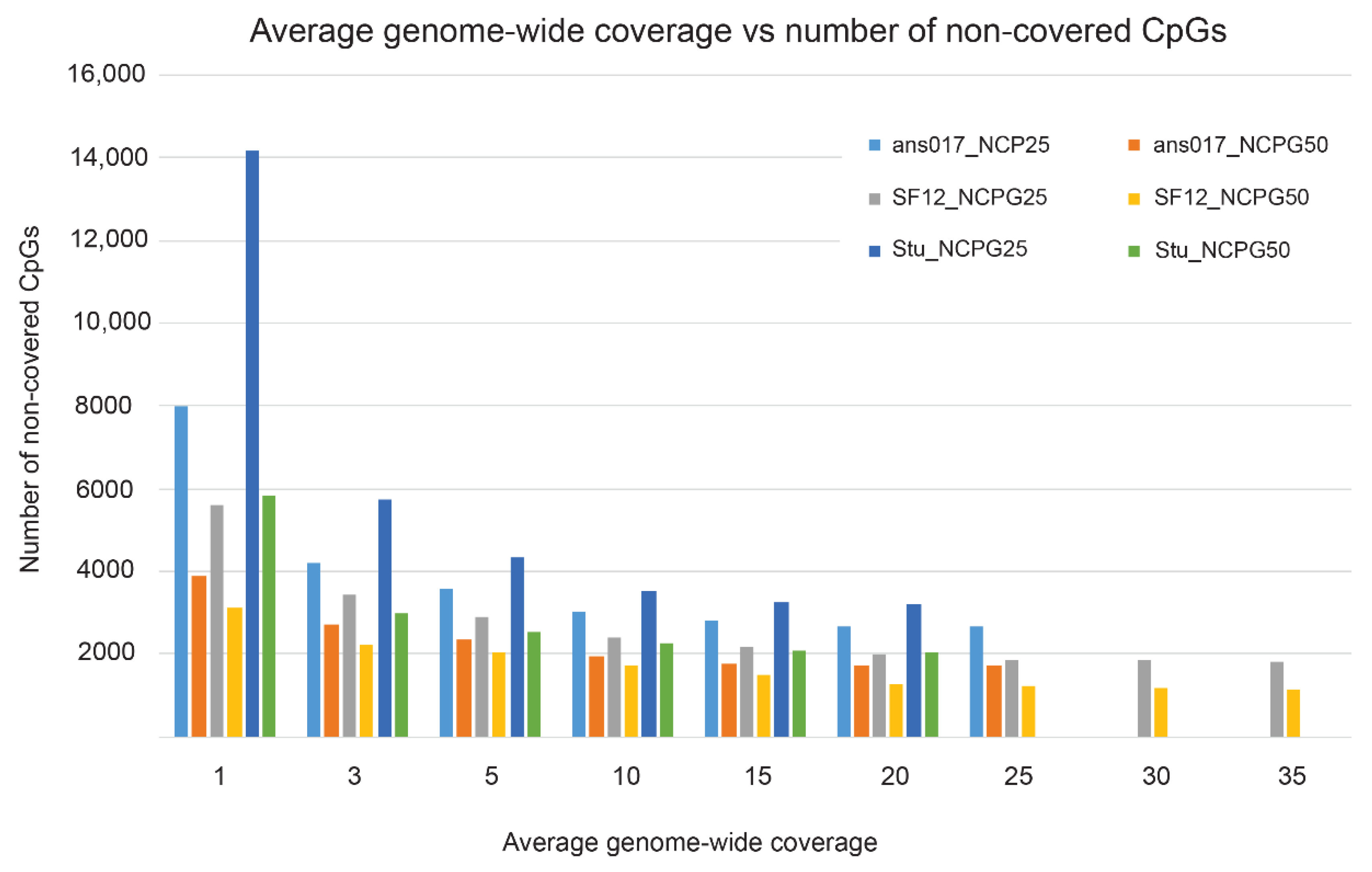
3.3. Practical Paleoepigenomic Considerations—The Importance of Sequencing Depth
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Nagl, N.G.; Wang, X.; Patsialou, A.; Van Scoy, M.; Moran, E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007, 26, 752–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. 2009, 10, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Wani, A.A. Histone modifications: Crucial elements for damage response and chromatin restoration. J. Cell. Physiol. 2010, 223, 283–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barisic, D.; Stadler, M.B.; Iurlaro, M.; Schubeler, D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 2019, 569, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Brettingham-Moore, K.H.; Taberlay, P.C.; Holloway, A.F. Interplay between Transcription Factors and the Epigenome: Insight from the Role of RUNX1 in Leukemia. Front. Immunol. 2015, 6, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Ardura, A.; Zaiko, A.; Moran, P.; Planes, S.; Garcia-Vazquez, E. Epigenetic signatures of invasive status in populations of marine invertebrates. Sci. Rep. 2017, 7, 42193. [Google Scholar] [CrossRef] [PubMed]
- Chown, S.L.; Hodgins, K.A.; Griffin, P.C.; Oakeshott, J.G.; Byrne, M.; Hoffmann, A.A. Biological invasions, climate change and genomics. Evol. Appl. 2015, 8, 23–46. [Google Scholar] [CrossRef]
- Marin, P.; Genitoni, J.; Barloy, D.; Maury, S.; Gibert, P.; Ghalambor, C.K.; Vieira, C. Biological invasion: The influence of the hidden side of the (epi)genome. Funct. Ecol. 2020, 34, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.A.; Martin, M.D.; Nguyen, M.; Mendelson, T.C. Epigenetic divergence as a potential first step in darter speciation. Mol. Ecol. 2016, 25, 1883–1894. [Google Scholar] [CrossRef]
- Charlesworth, D.; Barton, N.H.; Charlesworth, B. The sources of adaptive variation. Proc. R. Soc. B Biol. Sci. 2017, 284. [Google Scholar] [CrossRef] [Green Version]
- Liebl, A.L.; Schrey, A.W.; Richards, C.L.; Martin, L.B. Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integr. Comp. Biol. 2013, 53, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.W.; Stenzel, U.; Meyer, M.; Krause, J.; Kircher, M.; Paabo, S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010, 38, e87. [Google Scholar] [CrossRef] [Green Version]
- Gokhman, D.; Lavi, E.; Prufer, K.; Fraga, M.F.; Riancho, J.A.; Kelso, J.; Paabo, S.; Meshorer, E.; Carmel, L. Reconstructing the DNA methylation maps of the Neandertal and the Denisovan. Science 2014, 344, 523–527. [Google Scholar] [CrossRef]
- Orlando, L.; Gilbert, M.T.; Willerslev, E. Reconstructing ancient genomes and epigenomes. Nat. Rev. 2015, 16, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.; Green, R.E.; Kircher, M.; Krause, J.; Patterson, N.; Durand, E.Y.; Viola, B.; Briggs, A.W.; Stenzel, U.; Johnson, P.L.; et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 2010, 468, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokhman, D.; Meshorer, E.; Carmel, L. Epigenetics: It’s Getting Old. Past Meets Future in Paleoepigenetics. Trends Ecol. Evol. 2016, 31, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prufer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokhman, D.; Mishol, N.; de Manuel, M.; de Juan, D.; Shuqrun, J.; Meshorer, E.; Marques-Bonet, T.; Rak, Y.; Carmel, L. Reconstructing Denisovan Anatomy Using DNA Methylation Maps. Cell 2020, 180, 601. [Google Scholar] [CrossRef] [Green Version]
- Gokhman, D.; Nissim-Rafinia, M.; Agranat-Tamir, L.; Housman, G.; Garcia-Perez, R.; Lizano, E.; Cheronet, O.; Mallick, S.; Nieves-Colon, M.A.; Li, H.; et al. Differential DNA methylation of vocal and facial anatomy genes in modern humans. Nat. Commun. 2020, 11, 1186–1189. [Google Scholar] [CrossRef] [Green Version]
- Hanghoj, K.; Seguin-Orlando, A.; Schubert, M.; Madsen, T.; Pedersen, J.S.; Willerslev, E.; Orlando, L. Fast, Accurate and Automatic Ancient Nucleosome and Methylation Maps with epiPALEOMIX. Mol. Biol. Evol. 2016, 33, 3284–3298. [Google Scholar] [CrossRef] [Green Version]
- Hanghoj, K.; Renaud, G.; Albrechtsen, A.; Orlando, L. DamMet: Ancient methylome mapping accounting for errors, true variants, and post-mortem DNA damage. Gigascience 2019, 8, giz025. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.S.; Valen, E.; Velazquez, A.M.; Parker, B.J.; Rasmussen, M.; Lindgreen, S.; Lilje, B.; Tobin, D.J.; Kelly, T.K.; Vang, S.; et al. Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 2014, 24, 454–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.W.; Monroe, C.; Bolnick, D.A. Detection of Cytosine methylation in ancient DNA from five native american populations using bisulfite sequencing. PLoS ONE 2015, 10, e0125344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Redfern, A.D.; Colley, S.M.; Beveridge, D.J.; Ikeda, N.; Epis, M.R.; Li, X.; Foulds, C.E.; Stuart, L.M.; Barker, A.; Russell, V.J.; et al. RNA-induced silencing complex (RISC) Proteins PACT, TRBP, and Dicer are SRA binding nuclear receptor coregulators. Proc. Natl. Acad. Sci. USA 2013, 110, 6536–6541. [Google Scholar] [CrossRef] [Green Version]
- Buchon, N.; Vaury, C. RNAi: A defensive RNA-silencing against viruses and transposable elements. Heredity 2006, 96, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, H.; Casas-Mollano, J.A. On the origin and functions of RNA-mediated silencing: From protists to man. Curr. Genet. 2006, 50, 81–99. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, S.; Li, N.; Yang, X.; Ma, S.; Yang, A.; Zhang, H.; Yang, S.; Mao, C.; Xu, L.; et al. miR-34a Targets HDAC1-Regulated H3K9 Acetylation on Lipid Accumulation Induced by Homocysteine in Foam Cells. J. Cell. Biochem. 2017, 118, 4617–4627. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, Y.; Zhai, Y.; Ma, X.; An, X.; Zhang, S.; Li, Z. MicroRNA-29b regulates DNA methylation by targeting Dnmt3a/3b and Tet1/2/3 in porcine early embryo development. Dev. Growth Differ. 2018, 60, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Pitari, M.R.; Amodio, N.; Di Martino, M.T.; Conforti, F.; Leone, E.; Botta, C.; Paolino, F.M.; Del Giudice, T.; Iuliano, E.; et al. miR-29b negatively regulates human osteoclastic cell differentiation and function: Implications for the treatment of multiple myeloma-related bone disease. J. Cell. Physiol. 2013, 228, 1506–1515. [Google Scholar] [CrossRef]
- Llorens, F.; Thune, K.; Marti, E.; Kanata, E.; Dafou, D.; Diaz-Lucena, D.; Vivancos, A.; Shomroni, O.; Zafar, S.; Schmitz, M.; et al. Regional and subtype-dependent miRNA signatures in sporadic Creutzfeldt-Jakob disease are accompanied by alterations in miRNA silencing machinery and biogenesis. PLoS Pathog. 2018, 14, e1006802. [Google Scholar] [CrossRef] [Green Version]
- Loher, P.; Karathanasis, N.; Londin, E.; Bray, P.; Pliatsika, V.; Telonis, A.G.; Rigoutsos, I. IsoMiRmap-fast, deterministic, and exhaustive mining of isomiRs from short RNA-seq datasets. Bioinformatics 2021, 37, 1828–1838. [Google Scholar] [CrossRef]
- Keller, A.; Kreis, S.; Leidinger, P.; Maixner, F.; Ludwig, N.; Backes, C.; Galata, V.; Guerriero, G.; Fehlmann, T.; Franke, A.; et al. miRNAs in Ancient Tissue Specimens of the Tyrolean Iceman. Mol. Biol. Evol. 2017, 34, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Smith, O.; Dunshea, G.; Sinding, M.S.; Fedorov, S.; Germonpre, M.; Bocherens, H.; Gilbert, M.T.P. Ancient RNA from Late Pleistocene permafrost and historical canids shows tissue-specific transcriptome survival. PLoS Biol. 2019, 17, e3000166. [Google Scholar] [CrossRef] [Green Version]
- Fromm, B.; Tarbier, M.; Smith, O.; Marmol-Sanchez, E.; Dalen, L.; Gilbert, T.P.; Friedlander, M.R. Ancient microRNA profiles of a 14,300-year-old canid samples confirm taxonomic origin and give glimpses into tissue-specific gene regulation from the Pleistocene. RNA 2020, 27, 324–334. [Google Scholar] [CrossRef]
- Marino-Ramirez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteomics 2005, 2, 719–729. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Arents, G.; Moudrianakis, E.N. The histone fold: A ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc. Natl. Acad. Sci. USA 1995, 92, 11170–11174. [Google Scholar] [CrossRef] [Green Version]
- Raisner, R.M.; Hartley, P.D.; Meneghini, M.D.; Bao, M.Z.; Liu, C.L.; Schreiber, S.L.; Rando, O.J.; Madhani, H.D. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 2005, 123, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, M.; Amrichova, J.; Parker, B.J.; Soboleva, T.A.; Jack, C.; Williams, R.; Huttley, G.A.; Tremethick, D.J. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat. Struct. Mol. Biol. 2012, 19, 1076–1083. [Google Scholar] [CrossRef]
- Ku, M.; Jaffe, J.D.; Koche, R.P.; Rheinbay, E.; Endoh, M.; Koseki, H.; Carr, S.A.; Bernstein, B.E. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol. 2012, 13, R85. [Google Scholar] [CrossRef] [Green Version]
- Sarcinella, E.; Zuzarte, P.C.; Lau, P.N.; Draker, R.; Cheung, P. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol. Cell. Biol. 2007, 27, 6457–6468. [Google Scholar] [CrossRef] [Green Version]
- Chervona, Y.; Arita, A.; Costa, M. Carcinogenic metals and the epigenome: Understanding the effect of nickel, arsenic, and chromium. Metallomics 2012, 4, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Kanthasamy, A.; Anantharam, V.; Sun, F.; Kanthasamy, A.G. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: Relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 2010, 77, 621–632. [Google Scholar] [CrossRef]
- Han, N.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; Trillsch, F. H3K4me3 Is a Potential Mediator for Antiproliferative Effects of Calcitriol (1alpha,25(OH)2D3) in Ovarian Cancer Biology. Int. J. Mol. Sci. 2020, 21, 2151. [Google Scholar] [CrossRef] [Green Version]
- Mondal, P.; Sen, S.; Klein, B.J.; Tiwary, N.; Gadad, S.S.; Kutateladze, T.G.; Roy, S.; Das, C. TCF19 Promotes Cell Proliferation through Binding to the Histone H3K4me3 Mark. Biochemistry 2020, 59, 389–399. [Google Scholar] [CrossRef]
- Pena, P.V.; Hom, R.A.; Hung, T.; Lin, H.; Kuo, A.J.; Wong, R.P.; Subach, O.M.; Champagne, K.S.; Zhao, R.; Verkhusha, V.V.; et al. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J. Mol. Biol. 2008, 380, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Montavon, T.; Duboule, D. Chromatin organization and global regulation of HOX gene clusters. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120367. [Google Scholar] [CrossRef] [Green Version]
- Okitsu, C.Y.; Hsieh, C.L. DNA methylation dictates histone H3K4 methylation. Mol. Cell. Biol. 2007, 27, 2746–2757. [Google Scholar] [CrossRef] [Green Version]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Valouev, A.; Johnson, S.M.; Boyd, S.D.; Smith, C.L.; Fire, A.Z.; Sidow, A. Determinants of nucleosome organization in primary human cells. Nature 2011, 474, 516–520. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Bird, A.P. CpG islands as gene markers in the vertebrate nucleus. Trends Genet. 1987, 3, 342–347. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Curradi, M.; Izzo, A.; Badaracco, G.; Landsberger, N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 2002, 22, 3157–3173. [Google Scholar] [CrossRef] [Green Version]
- Heard, E.; Clerc, P.; Avner, P. X-chromosome inactivation in mammals. Annu. Rev. Genet. 1997, 31, 571–610. [Google Scholar] [CrossRef]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- Webster, M.J.; Knable, M.B.; O’Grady, J.; Orthmann, J.; Weickert, C.S. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol. Psychiatry 2002, 7, 924–985. [Google Scholar] [CrossRef] [Green Version]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonte, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.H.; Connelly, J.J.; Reilly, J.L.; Carter, C.S.; Drogos, L.L.; Pournajafi-Nazarloo, H.; Ruocco, A.C.; Keedy, S.K.; Matthew, I.; Tandon, N.; et al. Sex and diagnosis specific associations between DNA methylation of the oxytocin receptor gene with emotion processing and temporal-limbic and prefrontal brain volumes in psychotic disorders. Biol. Psychiatry. Cognitive Neurosci. Neuroimaging 2016, 1, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, N.B.; Richey, J.A.; Zvolensky, M.J.; Maner, J.K. Exploring human freeze responses to a threat stressor. J. Behav. Ther. Exp. Psychiatry 2008, 39, 292–304. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Giuliani, C.; Sazzini, M.; Bacalini, M.G.; Pirazzini, C.; Marasco, E.; Fontanesi, E.; Franceschi, C.; Luiselli, D.; Garagnani, P. Epigenetic Variability across Human Populations: A Focus on DNA Methylation Profiles of the KRTCAP3, MAD1L1 and BRSK2 Genes. Genome Biol. Evol. 2016, 8, 2760–2773. [Google Scholar] [CrossRef] [Green Version]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philos. Trans. R. Soc. London Series B Biol. Sci. 2009, 364, 3403–3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyn, H.; Moran, S.; Hernando-Herraez, I.; Sayols, S.; Gomez, A.; Sandoval, J.; Monk, D.; Hata, K.; Marques-Bonet, T.; Wang, L.; et al. DNA methylation contributes to natural human variation. Genome Res. 2013, 23, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Lumey, L.H.; Stein, A.D.; Susser, E. Prenatal famine and adult health. Annu. Rev. Public Health 2011, 32, 237–262. [Google Scholar] [CrossRef] [Green Version]
- Painter, R.C.; Osmond, C.; Gluckman, P.; Hanson, M.; Phillips, D.I.; Roseboom, T.J. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 2008, 115, 1243–1249. [Google Scholar] [CrossRef]
- Fagny, M.; Patin, E.; MacIsaac, J.L.; Rotival, M.; Flutre, T.; Jones, M.J.; Siddle, K.J.; Quach, H.; Harmant, C.; McEwen, L.M.; et al. The epigenomic landscape of African rainforest hunter-gatherers and farmers. Nat. Commun. 2015, 6, 10047. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Salas, P.; Moore, S.E.; Baker, M.S.; Bergen, A.W.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Guan, Y.; Laritsky, E.; Silver, M.J.; et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat. Commun. 2014, 5, 3746. [Google Scholar] [CrossRef] [PubMed]
- Geisel, J.; Schorr, H.; Bodis, M.; Isber, S.; Hubner, U.; Knapp, J.P.; Obeid, R.; Herrmann, W. The vegetarian lifestyle and DNA methylation. Clin. Chem. Lab. Med. 2005, 43, 1164–1169. [Google Scholar] [CrossRef]
- Sebastiani, G.; Herranz Barbero, A.; Borras-Novell, C.; Alsina Casanova, M.; Aldecoa-Bilbao, V.; Andreu-Fernandez, V.; Pascual Tutusaus, M.; Ferrero Martinez, S.; Gomez Roig, M.D.; Garcia-Algar, O. The Effects of Vegetarian and Vegan Diet during Pregnancy on the Health of Mothers and Offspring. Nutrients 2019, 11, 557. [Google Scholar] [CrossRef] [Green Version]
- Gadgil, M.S.; Joshi, K.S.; Naik, S.S.; Pandit, A.N.; Otiv, S.R.; Patwardhan, B.K. Association of homocysteine with global DNA methylation in vegetarian Indian pregnant women and neonatal birth anthropometrics. J. Matern. Fetal. Neonatal Med. 2014, 27, 1749–1753. [Google Scholar] [CrossRef]
- Waterland, R.A.; Kellermayer, R.; Laritsky, E.; Rayco-Solon, P.; Harris, R.A.; Travisano, M.; Zhang, W.; Torskaya, M.S.; Zhang, J.; Shen, L.; et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010, 6, e1001252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, C.J.; Fuller, N.J.; Prentice, A.M. Folate status during pregnancy and lactation in a West African rural community. Hum. Nutr. Nutr. 1986, 40, 3–13. [Google Scholar]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Borghol, N.; Suderman, M.; McArdle, W.; Racine, A.; Hallett, M.; Pembrey, M.; Hertzman, C.; Power, C.; Szyf, M. Associations with early-life socio-economic position in adult DNA methylation. Int. J. Epidemiol. 2012, 41, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Laubach, Z.M.; Perng, W.; Cardenas, A.; Rifas-Shiman, S.L.; Oken, E.; Demeo, D.; Litonjua, A.A.; Duca, R.-C.; Godderis, L.; Baccarelli, A.; et al. Socioeconomic status and DNA methylation from birth through mid-childhood: A prospective study in Project Viva. Epigenomics 2019, 11, 1413–1427. [Google Scholar] [CrossRef]
- Fiorito, G.; McCrory, C.; Robinson, O.; Carmeli, C.; Rosales, C.O.; Zhang, Y.; Colicino, E.; Dugué, P.-A.; Artaud, F.; McKay, G.J.; et al. Socioeconomic position, lifestyle habits and biomarkers of epigenetic aging: A multi-cohort analysis. Aging 2019, 11, 2045–2070. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.L.; Smith, J.A.; Zhao, W.; Wang, X.; Mukherjee, B.; Kardia, S.L.; Shively, C.A.; Seeman, T.E.; Liu, Y.; Diez Roux, A. V Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: The multi-ethnic study of atherosclerosis. Epigenetics 2015, 10, 958–969. [Google Scholar] [CrossRef] [Green Version]
- Unternaehrer, E.; Luers, P.; Mill, J.; Dempster, E.; Meyer, A.H.; Staehli, S.; Lieb, R.; Hellhammer, D.H.; Meinlschmidt, G. Dynamic changes in DNA methylation of stress-associated genes (OXTR, BDNF ) after acute psychosocial stress. Transl. Psychiatry 2012, 2, e150. [Google Scholar] [CrossRef] [Green Version]
- Galanter, J.M.; Gignoux, C.R.; Oh, S.S.; Torgerson, D.; Pino-Yanes, M.; Thakur, N.; Eng, C.; Hu, D.; Huntsman, S.; Farber, H.J.; et al. Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures. eLife 2017, 6, e20532. [Google Scholar] [CrossRef]
- Sahm, A.; Koch, P.; Horvath, S.; Hoffmann, S. An Analysis of Methylome Evolution in Primates. Mol. Biol. Evol. 2021, 38, 4700–4714. [Google Scholar] [CrossRef]
- Gokhman, D.; Malul, A.; Carmel, L. Inferring Past Environments from Ancient Epigenomes. Mol. Biol. Evol. 2017, 34, 2429–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.W.; Non, A.L. Assessing the achievements and uncertain future of paleoepigenomics. Epigenomics 2021. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Bowman, B.; Freiberger, M.; Ryder, O.A.; Wilson, A.C. DNA sequences from the quagga, an extinct member of the horse family. Nature 1984, 312, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Llamas, B.; Holland, M.L.; Chen, K.; Cropley, J.E.; Cooper, A.; Suter, C.M. High-resolution analysis of cytosine methylation in ancient DNA. PLoS ONE 2012, 7, e30226. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do it right or not at all. Science 2000, 289, 1139. [Google Scholar] [CrossRef]
- Hofreiter, M.; Serre, D.; Poinar, H.N.; Kuch, M.; Paabo, S. Ancient DNA. Nat. Rev. 2001, 2, 353–359. [Google Scholar] [CrossRef]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Paabo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef]
- Bouchet, F.; Guidon, N.; Dittmar, K.; Harter, S.; Ferreira, L.F.; Chaves, S.M.; Reinhard, K.; Araujo, A. Parasite remains in archaeological sites. Mem. Inst. Oswaldo Cruz 2003, 98 (Suppl. 1), 47–52. [Google Scholar] [CrossRef]
- Knapp, M.; Clarke, A.C.; Horsburgh, K.A.; Matisoo-Smith, E.A. Setting the stage—building and working in an ancient DNA laboratory. Ann. Anat. 2012, 194, 3–6. [Google Scholar] [CrossRef]
- Hoss, M.; Jaruga, P.; Zastawny, T.H.; Dizdaroglu, M.; Paabo, S. DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res. 1996, 24, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Karlstrom, O. Heat-induced depyrimidination of deoxyribonucleic acid in neutral solution. Biochemistry 1973, 12, 5151–5154. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.; Krause, J.; Guschanski, K.; Savolainen, V.; Paabo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 2012, 7, e34131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiller, M.; Green, R.E.; Ronan, M.; Simons, J.F.; Du, L.; He, W.; Egholm, M.; Rothberg, J.M.; Keates, S.G.; Ovodov, N.D.; et al. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 13578–13584. [Google Scholar] [CrossRef] [Green Version]
- Hansen, A.; Willerslev, E.; Wiuf, C.; Mourier, T.; Arctander, P. Statistical evidence for miscoding lesions in ancient DNA templates. Mol. Biol. Evol. 2001, 18, 262–265. [Google Scholar] [CrossRef] [Green Version]
- Paabo, S. Ancient DNA: Extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 1939–1943. [Google Scholar] [CrossRef] [Green Version]
- Rubi, T.L.; Knowles, L.L.; Dantzer, B. Museum epigenomics: Characterizing cytosine methylation in historic museum specimens. Mol. Ecol. Resour. 2020, 20, 1161–1170. [Google Scholar] [CrossRef]
- Seguin-Orlando, A.; Hoover, C.A.; Vasiliev, S.K.; Ovodov, N.D.; Shapiro, B.; Cooper, A.; Rubin, E.M.; Willerslev, E.; Orlando, L. Amplification of TruSeq ancient DNA libraries with AccuPrime Pfx: Consequences on nucleotide misincorporation and methylation patterns. Sci. Technol. Archaeol. Res. 2015, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Seguin-Orlando, A.; Gamba, C.; Sarkissian, C.; Ermini, L.; Louvel, G.; Boulygina, E.; Sokolov, A.; Nedoluzhko, A.; Lorenzen, E.D.; Lopez, P.; et al. Pros and cons of methylation-based enrichment methods for ancient DNA. Sci. Rep. 2015, 5, 11826. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tollefsbol, T.O. DNA methylation detection: Bisulfite genomic sequencing analysis. Methods Mol. Biol. 2011, 791, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Hatahet, Z.; Melamede, R.J.; Kow, Y.W.; Wallace, S.S. Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem. 1997, 272, 32230–32239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakyan, V.K.; Blewitt, M.E.; Druker, R.; Preis, J.I.; Whitelaw, E. Metastable epialleles in mammals. Trends Genet. 2002, 18, 348–351. [Google Scholar] [CrossRef]
- Roemer, I.; Reik, W.; Dean, W.; Klose, J. Epigenetic inheritance in the mouse. Curr. Biol. 1997, 7, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.R.; Urich, M.A.; Chen, H.; et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef]
- Hatala, A.R.; Desjardins, M.; Bombay, A. Reframing Narratives of Aboriginal Health Inequity: Exploring Cree Elder Resilience and Well-Being in Contexts of Historical Trauma. Qual. Health Res. 2016, 26, 1911–1927. [Google Scholar] [CrossRef]
- Kowal, E.; Warin, M. Anthropology, Indigeneity, and the Epigenome: Special Section on Medical Anthropology. Am. Anthropol. 2018, 120, 822–825. [Google Scholar] [CrossRef]
- Wagner, J.K.; Colwell, C.; Claw, K.G.; Stone, A.C.; Bolnick, D.A.; Hawks, J.; Brothers, K.B.; Garrison, N.A. Fostering Responsible Research on Ancient DNA. Am. J. Hum. Genet. 2020, 107, 183–195. [Google Scholar] [CrossRef]
- Liu, Y.; Weyrich, L.S.; Llamas, B. More Arrows in the Ancient DNA Quiver: Use of Paleoepigenomes and Paleomicrobiomes to Investigate Animal Adaptation to Environment. Mol. Biol. Evol. 2020, 37, 307–319. [Google Scholar] [CrossRef]
- Seguin-Orlando, A.; Donat, R.; Der Sarkissian, C.; Southon, J.; Thèves, C.; Manen, C.; Tchérémissinoff, Y.; Crubézy, E.; Shapiro, B.; Deleuze, J.-F.; et al. Heterogeneous Hunter-Gatherer and Steppe-Related Ancestries in Late Neolithic and Bell Beaker Genomes from Present-Day France. Curr. Biol. 2021, 31, 1072–1083.e10. [Google Scholar] [CrossRef]
- Zakany, J.; Duboule, D. The role of HOX genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007, 17, 359–366. [Google Scholar] [CrossRef]
- Weaver, T.D. The meaning of Neandertal skeletal morphology. Proc. Natl. Acad. Sci. USA 2009, 106, 16028–16033. [Google Scholar] [CrossRef] [Green Version]
- Kohler, S.; Doelken, S.C.; Mungall, C.J.; Bauer, S.; Firth, H.V.; Bailleul-Forestier, I.; Black, G.C.; Brown, D.L.; Brudno, M.; Campbell, J.; et al. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014, 42, 966. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Welker, F.; Shen, C.C.; Bailey, S.E.; Bergmann, I.; Davis, S.; Xia, H.; Wang, H.; Fischer, R.; Freidline, S.E.; et al. A late Middle Pleistocene Denisovan mandible from the Tibetan Plateau. Nature 2019, 569, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Kircher, M.; Gansauge, M.T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prufer, K.; de Filippo, C.; et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Prufer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; de Filippo, C.; et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Malmstrom, H.; Omrak, A.; Raghavan, M.; Valdiosera, C.; Gunther, T.; Hall, P.; Tambets, K.; Parik, J.; Sjogren, K.G.; et al. Genomic diversity and admixture differs for Stone-Age Scandinavian foragers and farmers. Science 2014, 344, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Valdiosera, C.; Malmstrom, H.; Urena, I.; Rodriguez-Varela, R.; Sverrisdottir, O.O.; Daskalaki, E.A.; Skoglund, P.; Naidoo, T.; Svensson, E.M.; et al. Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc. Natl. Acad. Sci. USA 2015, 112, 11917–11922. [Google Scholar] [CrossRef] [Green Version]
- Gunther, T.; Malmstrom, H.; Svensson, E.M.; Omrak, A.; Sanchez-Quinto, F.; Kilinc, G.M.; Krzewinska, M.; Eriksson, G.; Fraser, M.; Edlund, H.; et al. Population genomics of Mesolithic Scandinavia: Investigating early postglacial migration routes and high-latitude adaptation. PLoS Biol. 2018, 16, e2003703. [Google Scholar] [CrossRef]
- Lazaridis, I.; Patterson, N.; Mittnik, A.; Renaud, G.; Mallick, S.; Kirsanow, K.; Sudmant, P.H.; Schraiber, J.G.; Castellano, S.; Lipson, M.; et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 2014, 513, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Fraser, M.; Sánchez Quinto, F. Human evolution. Department of Organismal biology, Uppsala University, Norbyvägen 18c, 75236 Uppsala, Sweden and Instituto Nacional de Medicina Genómica, 14610 Mexico City, Mexico. 2022; manuscript in preparation. [Google Scholar]
- Fraser, M. People of the Dolmens and Stone Cists: An Archaeogenetic investigation of Megalithic Graves from the Neolithic Period on Gotland. Doctoral Thesis, Uppsala University-Campus Gotland, Uppsala, Sweden, 2018. [Google Scholar]
- Schubert, M.; Ermini, L.; Der Sarkissian, C.; Jonsson, H.; Ginolhac, A.; Schaefer, R.; Martin, M.D.; Fernandez, R.; Kircher, M.; McCue, M.; et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014, 9, 1056–1082. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup, 1000 Genome Project Data Processing the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.R.; Wegmann, D.; Ehm, M.G.; Kessner, D.; St Jean, P.; Verzilli, C.; Shen, J.; Tang, Z.; Bacanu, S.A.; Fraser, D.; et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 2012, 337, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kuhnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. eLife 2018, 7, e36666. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, V.J.; Avanzi, C.; Krause-Kyora, B.; Seitz, A.; Herbig, A.; Inskip, S.; Bonazzi, M.; Reiter, E.; Urban, C.; Dangvard Pedersen, D.; et al. Ancient genomes reveal a high diversity of Mycobacterium leprae in medieval Europe. PLoS Pathog. 2018, 14, e1006997. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niiranen, L.; Leciej, D.; Edlund, H.; Bernhardsson, C.; Fraser, M.; Quinto, F.S.; Herzig, K.-H.; Jakobsson, M.; Walkowiak, J.; Thalmann, O. Epigenomic Modifications in Modern and Ancient Genomes. Genes 2022, 13, 178. https://doi.org/10.3390/genes13020178
Niiranen L, Leciej D, Edlund H, Bernhardsson C, Fraser M, Quinto FS, Herzig K-H, Jakobsson M, Walkowiak J, Thalmann O. Epigenomic Modifications in Modern and Ancient Genomes. Genes. 2022; 13(2):178. https://doi.org/10.3390/genes13020178
Chicago/Turabian StyleNiiranen, Laura, Dawid Leciej, Hanna Edlund, Carolina Bernhardsson, Magdalena Fraser, Federico Sánchez Quinto, Karl-Heinz Herzig, Mattias Jakobsson, Jarosław Walkowiak, and Olaf Thalmann. 2022. "Epigenomic Modifications in Modern and Ancient Genomes" Genes 13, no. 2: 178. https://doi.org/10.3390/genes13020178