
Identification of a Novel Epithelial–Mesenchymal Transition-Related Gene Signature for Endometrial Carcinoma Prognosis


Abstract
:1. Introduction
2. Method
2.1. Data Preparation
2.2. Differentially Expressed ERGs
2.3. Enrichment Analysis of Intersection Genes
2.4. Individualized Gene Signature
2.5. Nomogram Construction
2.6. Tumor Immune Microenvironment Analysis
2.7. Immunotherapy and Chemotherapy Response Prediction
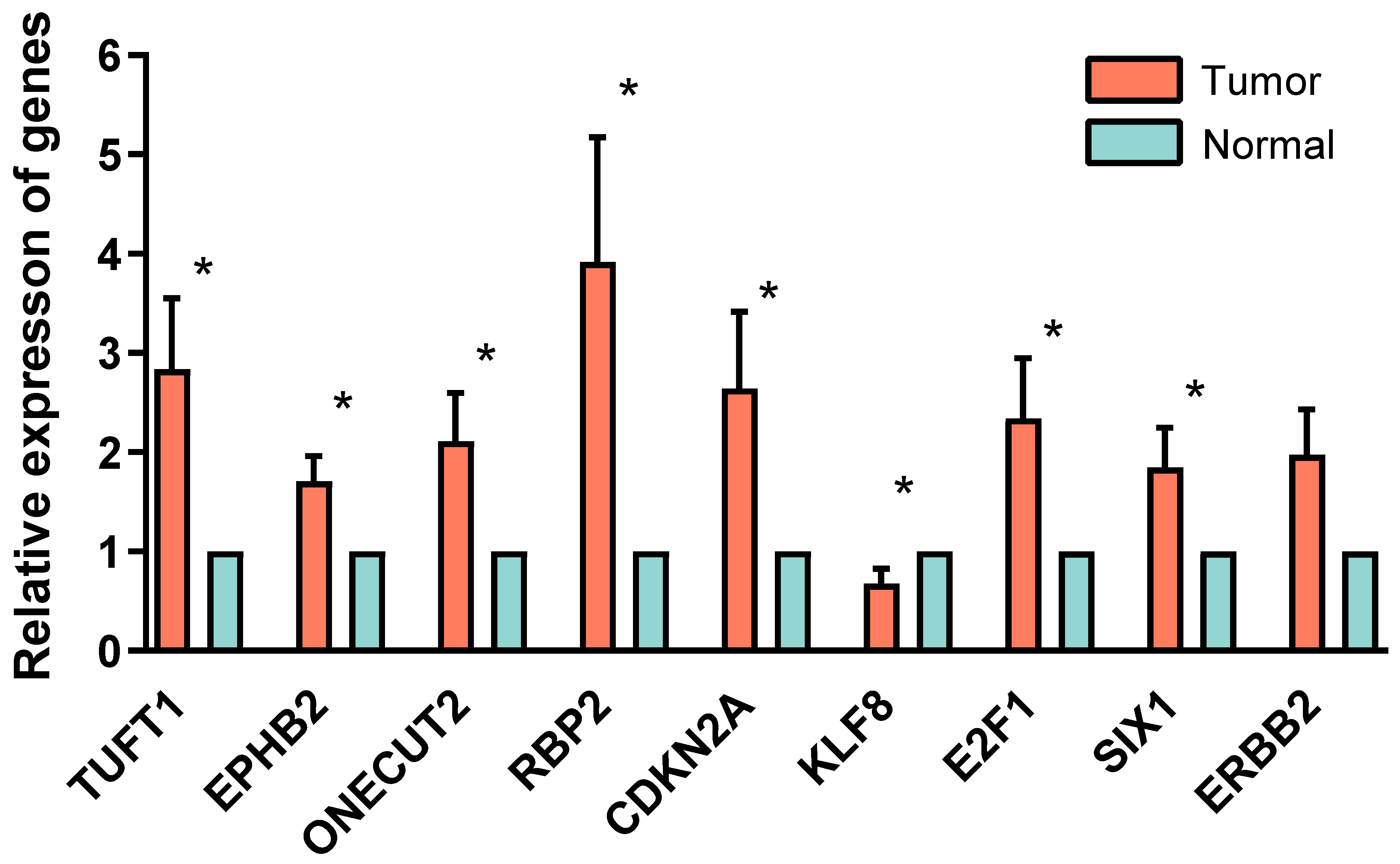
2.8. PCR Validation
3. Result
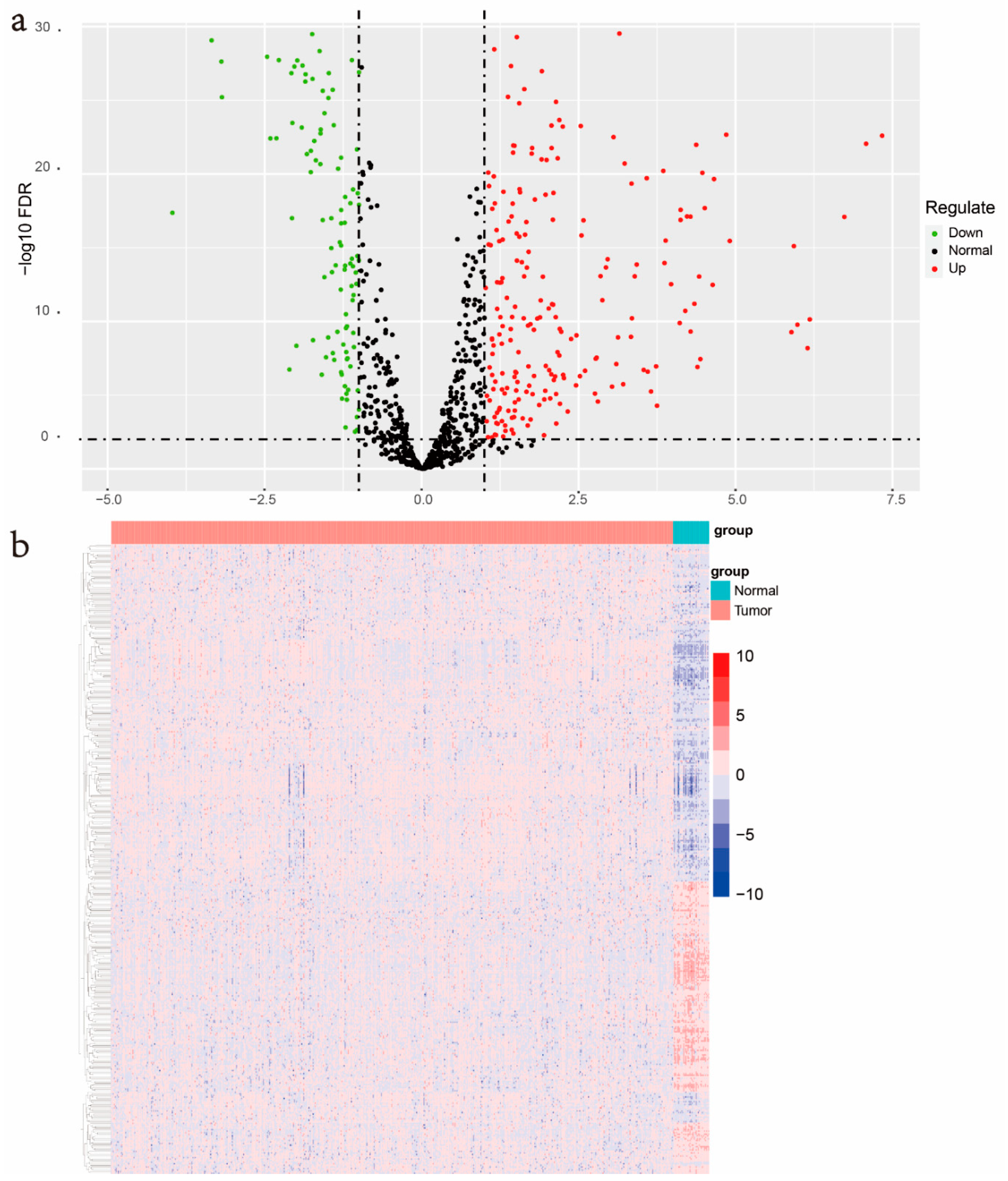
3.1. Identification of Differentially Expressed ERGs
3.2. Biological Functions Pathways Analysis
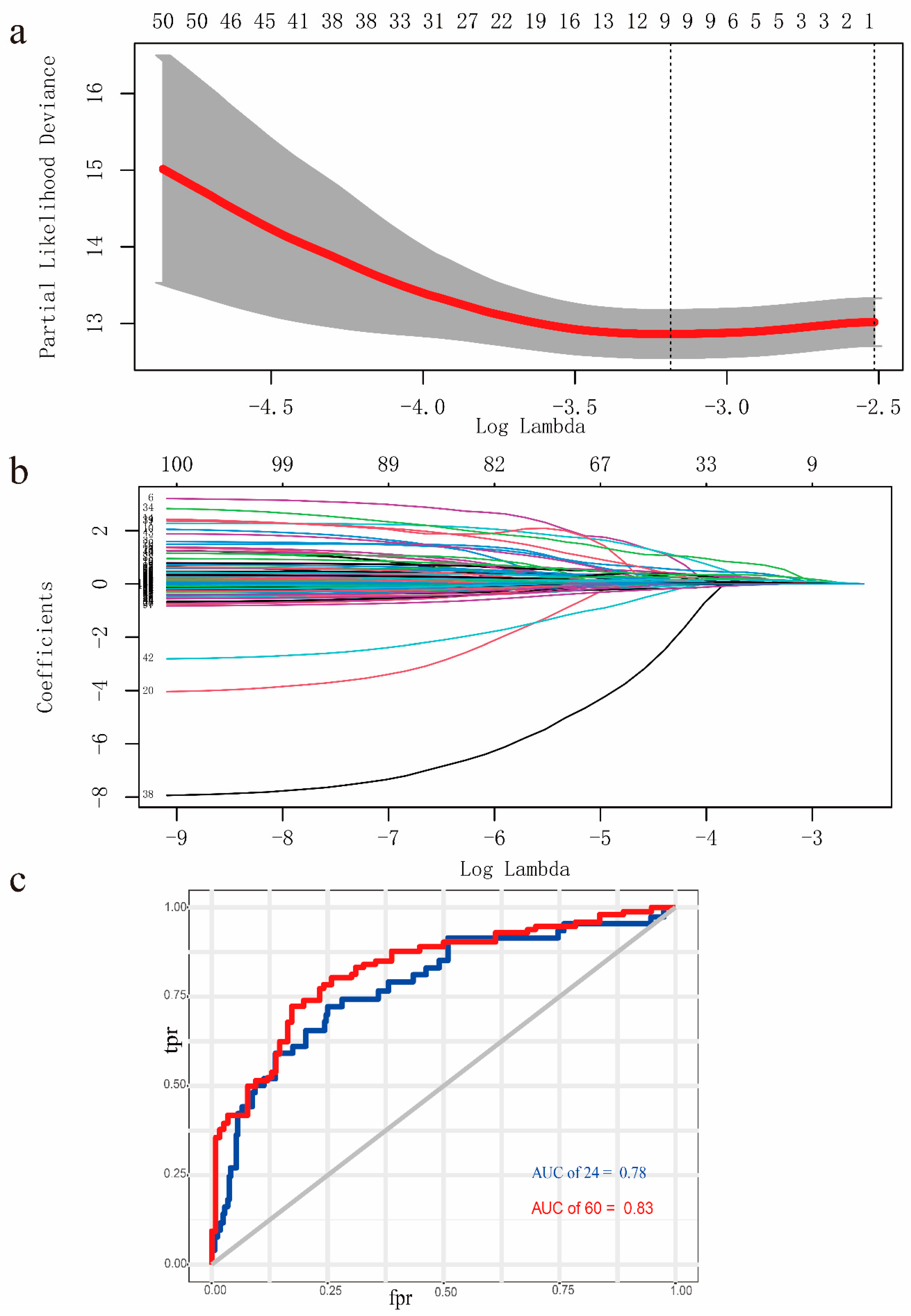
3.3. Screened for the ERGs with Significant Prognosis
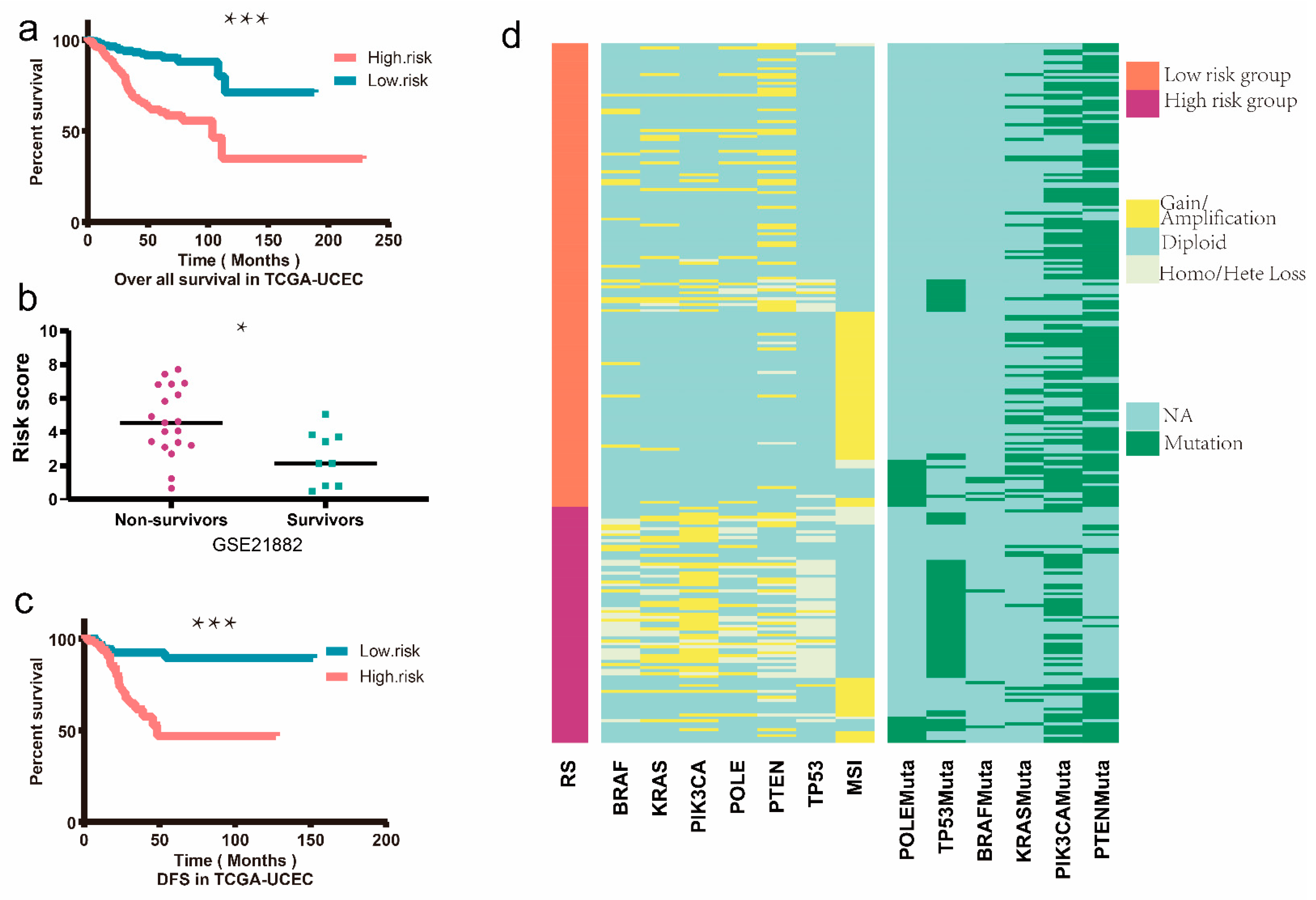
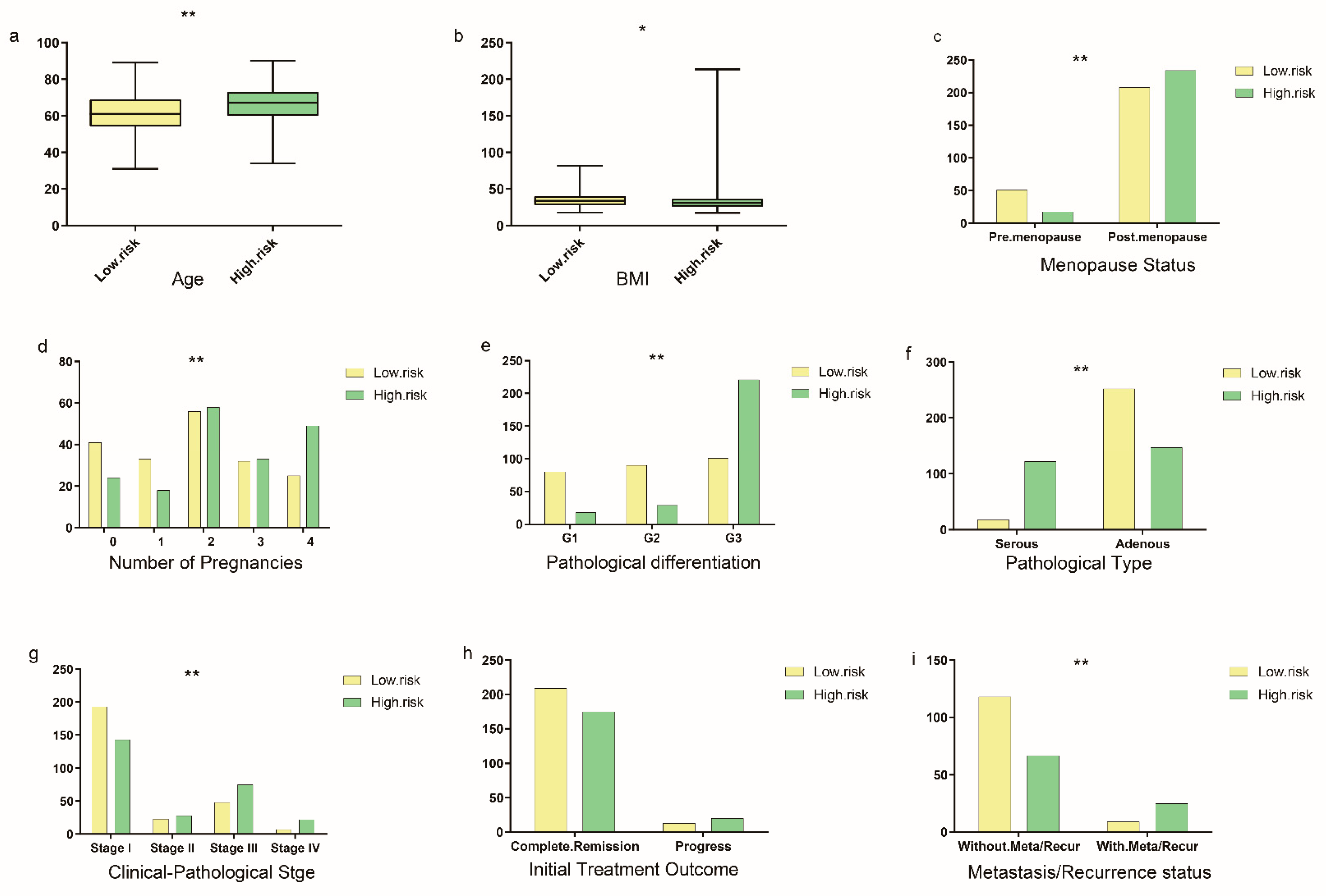
3.4. Establishment and Validation of the Prognostic EMT-Related Gene Signature
3.5. Nomogram Construction and Validation
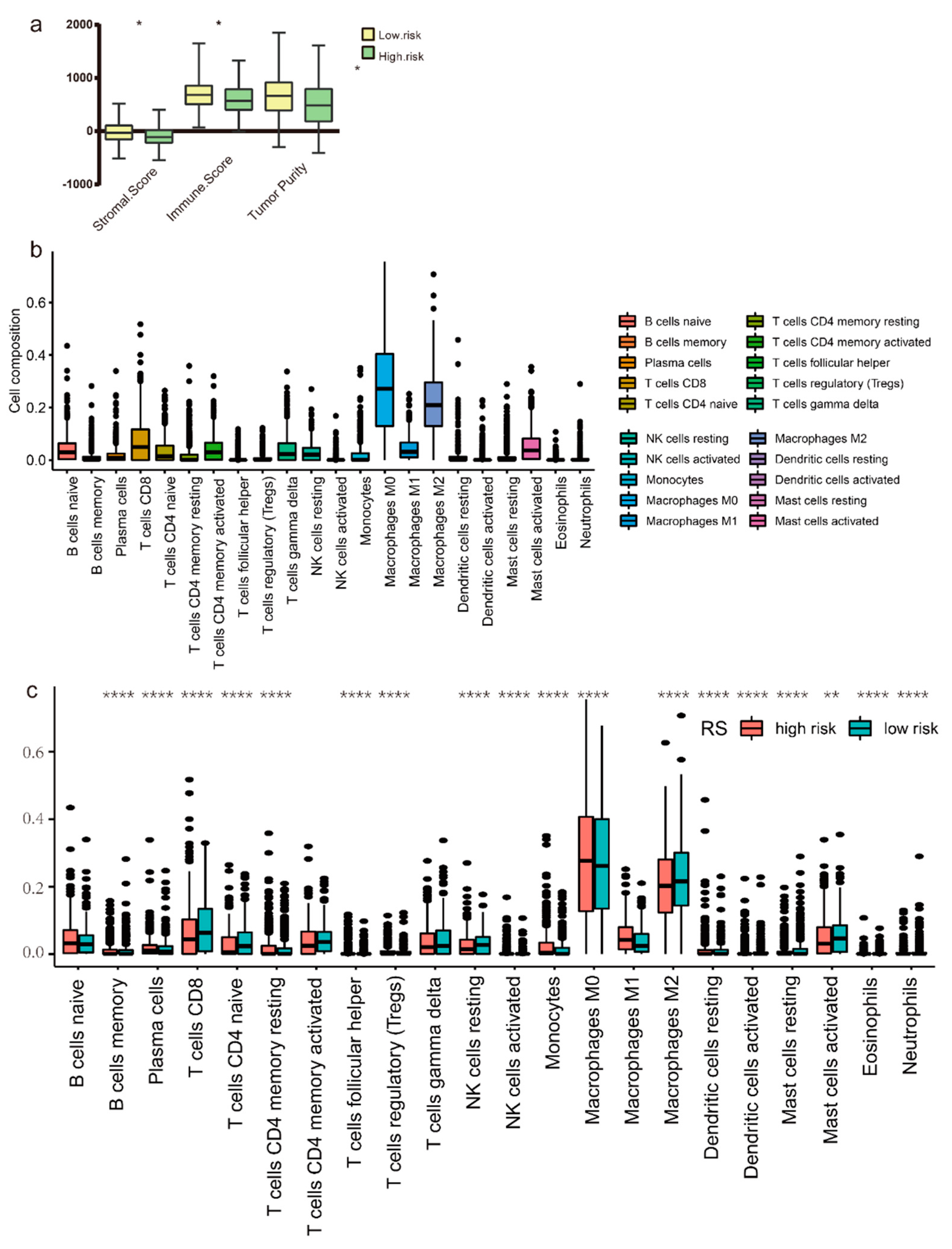
3.6. Immune Microenvironment Analysis
3.7. Immune Checkpoint Blockades Therapy and Drug Sensitivity Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EC | endometrial cancer |
| EMT | epithelial—mesenchymal transition |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| EPHB2 | EPH receptor B2 |
| TUFT1 | tuftelin 1 |
| CDKN2A | cyclin-dependent kinase inhibitor 2A |
| ONECUT2 | one cut homeobox 2 |
| RBP2 | retinol-binding protein 2 |
| KLF8 | Kruppel-like factor 8 |
| E2F1 | E2F transcription factor 1 |
| SIX1 | SIX homeobox 1 |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 |
| RS | risk score |
| ESTIMATE | estimation of stromal and immune cells in malignant tumor tissues using expression data |
| ICB | immune checkpoint blockade |
| TIDE | tumor immune dysfunction and exclusion |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| ERGs | EMT-related genes |
| UCEC | uterine corpus endometrial carcinoma |
| ROC | receiver-operating characteristic |
| TME | tumor microenvironment |
| LASSO | least absolute shrinkage and selection operator |
| IC50 | the half-maximal inhibitory concentration |
| FDR | false discovery rate |
| MMRd | mismatch repair-deficient |
| CTLA-4 | cytotoxic T-lymphocyte antigen 4 |
| PD-L1 | programmed cell death-ligand 1 |
| FIGO | The International Federation of Gynecology and Obstetrics |
| AUC | area under the curve |
| OS | overall survival |
| DFS | disease-free survival |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat. Commun. 2019, 10, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Lim, C.T. Tumor dissemination: An EMT affair. Cancer Cell 2013, 23, 272–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.C.; Li, C.J.; Yiang, G.T.; Tsai, A.P.; Wu, M.Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. J. Clin. Med. 2019, 8, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, E.; Caserta, C.A.; Rumio, C.; Marcucci, F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells 2019, 8, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, P.; Chen, L.; Zhao, L.; Zhu, J.; Zhu, T. The Long Non-Coding RNA-14327.1 Promotes Migration and Invasion Potential of Endometrial Carcinoma Cells by Stabilizing the Potassium Channel Kca3.1. Onco Targets Ther. 2019, 12, 10287–10297. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wickham, H.; Sievert, C. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: Houston, TX, USA, 2016. [Google Scholar]
- Slowikowski, K.; Schep, A.; Hughes, S.; Dang, T.K. Ggrepel: Automatically Position Non-Overlapping Text Labels with ‘Ggplot2’. 2020. Available online: https://rdrr.io/cran/ggrepel/ (accessed on 29 October 2020).
- Perry, M. Heatmaps: Flexible Heatmaps for Functional Genomics and Sequence Features. 2020. Available online: https://rdrr.io/bioc/heatmaps/ (accessed on 29 October 2020).
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenf Uhrer, J. Enrichment Analysis for Gene Ontology. 2006. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 29 October 2020).
- Weijun, L.; Cory, B.J.B. Pathview: An R/Bioconductor Package for Pathway-Based Data Integration and Visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Gentry, J.; Long, L.; Gentleman, R.; Falcon, S. Provides Plotting Capabilities for R Graph Objects. 2007. Available online: https://bioconductor.org/packages/release/bioc/html/Rgraphviz.html (accessed on 29 October 2020).
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [PubMed]
- Iasonos, A.; Schrag, D.; Raj, G.V.; Panageas, K.S. How to build and interpret a nomogram for cancer prognosis. J. Clin. Oncol. 2008, 26, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression Shrinkage and Selection via the LASSO. J. R. Statist. Soc. 1996, 73, 273–282. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 2020, 12, 21. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Khan, H.; Siddiqui, Z.; Khan, M.Y.; Rehman, S.; Shahab, U.; Godovikova, T.; Silnikov, V. AGEs, RAGEs and s-RAGE; friend or foe for cancer. Semin Cancer Biol. 2018, 49, 44–55. [Google Scholar] [CrossRef]
- Waghela, B.N.; Vaidya, F.U.; Ranjan, K.; Chhipa, A.S.; Tiwari, B.S.; Pathak, C. AGE-RAGE synergy influences programmed cell death signaling to promote cancer. Mol. Cell Biochem. 2020, 476, 585–598. [Google Scholar] [CrossRef]
- Furlan, D.; Carnevali, I.; Marcomini, B.; Cerutti, R.; Dainese, E.; Capella, C.; Riva, C. The high frequency of de novo promoter methylation in synchronous primary endometrial and ovarian carcinomas. Clin. Cancer Res. 2006, 12, 3329–3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Wang, H.; Miao, J.; Liang, Y. Clinicopathological Significance and Potential Drug Target of CDKN2A/p16 in Endometrial Carcinoma. Sci. Rep. 2015, 5, 13238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousin, S.; Khalifa, E.; Crombe, A.; Laizet, Y.; Lucchesi, C.; Toulmonde, M.; Le Moulec, S.; Auzanneau, C.; Soubeyran, I.; Italiano, A. Targeting ERBB2 mutations in solid tumors: Biological and clinical implications. J. Hematol. Oncol. 2018, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Han, J.; Shi, S.; Dai, Y.; He, J. TUFT1 promotes metastasis and chemoresistance in triple negative breast cancer through the TUFT1/Rab5/Rac1 pathway. Cancer Cell Int. 2019, 19, 242. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.C.; Lee, C.F.; Li, Y.S.; Chen, Y.R.; Hsiao, P.W.; Chan, M.Y.; Lin, F.M.; Huang, H.D.; Chen, Y.T.; Jeng, Y.M.; et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, H.; Urvalek, A.M.; Li, T.; Yu, L.; Lamar, J.; DiPersio, C.M.; Feustel, P.J.; Zhao, J. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene 2011, 30, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Suen, A.A.; Jefferson, W.N.; Wood, C.E.; Padilla-Banks, E.; Bae-Jump, V.L.; Williams, C.J. SIX1 Oncoprotein as a Biomarker in a Model of Hormonal Carcinogenesis and in Human Endometrial Cancer. Mol. Cancer Res. 2016, 14, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Suen, A.A.; Jefferson, W.N.; Wood, C.E.; Williams, C.J. SIX1 Regulates Aberrant Endometrial Epithelial Cell Differentiation and Cancer Latency Following Developmental Estrogenic Chemical Exposure. Mol. Cancer Res. 2019, 17, 2369–2382. [Google Scholar] [CrossRef] [Green Version]
- Ghatalia, P.; Zibelman, M.; Geynisman, D.M.; Plimack, E.R. Checkpoint Inhibitors for the Treatment of Renal Cell Carcinoma. Curr. Treat Options Oncol. 2017, 18, 7. [Google Scholar] [CrossRef]
- Zhan, H.X.; Zhou, B.; Cheng, Y.G.; Xu, J.W.; Wang, L.; Zhang, G.Y.; Hu, S.Y. Crosstalk between stromal cells and cancer cells in pancreatic cancer: New insights into stromal biology. Cancer Lett. 2017, 392, 83–93. [Google Scholar] [CrossRef]
- Ma, H.Y.; Liu, X.Z.; Liang, C.M. Inflammatory microenvironment contributes to epithelial-mesenchymal transition in gastric cancer. World J. Gastroenterol. 2016, 22, 6619–6628. [Google Scholar] [CrossRef] [PubMed]
- Levan, K.; Partheen, K.; Osterberg, L.; Olsson, B.; Delle, U.; Eklind, S.; Horvath, G. Identification of a gene expression signature for survival prediction in type I endometrial carcinoma. Gene Expr. 2010, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lin, J.; He, H. Identification of Potential Crucial Genes Associated With the Pathogenesis and Prognosis of Endometrial Cancer. Front. Genet. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Oza, A.M.; Pignata, S.; Poveda, A.; McCormack, M.; Clamp, A.; Schwartz, B.; Cheng, J.; Li, X.; Campbell, K.; Dodion, P.; et al. Randomized Phase II Trial of Ridaforolimus in Advanced Endometrial Carcinoma. J. Clin. Oncol. 2015, 33, 3576–3582. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| EPHB2 Foward | AGAAACGCTAATGGACTCCACT |
| EPHB2 Reverse | GTGCGGATCGTGTTCATGTT |
| TUFT1 Foward | TCAGACTGTGTGGCTTTTGAG |
| TUFT1 Reverse | GTCAGCATTGTTGCTCCGAAG |
| CDKN2A Foward | GATCCAGGTGGGTAGAAGGTC |
| CDKN2A Reverse | CCCCTGCAAACTTCGTCCT |
| ONECUT2 Foward | GGAATCCAAAACCGTGGAGTAA |
| ONECUT2 Reverse | CTCTTTGCGTTTGCACGCTG |
| RBP2 Foward | TTTTGCCACCCGCAAGATTG |
| RBP2 Reverse | CGGAATGTGCTAGTGGTTTTTGT |
| KLF8 Foward | CCCAAGTGGAACCAGTTGACC |
| KLF8 Reverse | GACGTGGACACCACAAGGG |
| E2F1 Forward | ACGCTATGAGACCTCACTGAA |
| E2F1 Reverse | TCCTGGGTCAACCCCTCAAG |
| SIX1 Forward | CTGCCGTCGTTTGGCTTTAC |
| SIX1 Reverse | GCTCTCGTTCTTGTGCAGGT |
| ERBB2 Foward | TGCAGGGAAACCTGGAACTC |
| ERBB2 Reverse | ACAGGGGTGGTATTGTTCAGC |
| GAPDH Foward | AGATCCCTCCAAAATCAAGTGG |
| GAPDH Reverse | GGCAGAGATGATGACCCTTTT |
| Univariate Cox Regression | Multivariate Cox Regression | |||||
|---|---|---|---|---|---|---|
| Variates | p | HR | 95% CI | p | HR | 95% CI |
| Age | 0.001 | 1.038 | 1.017–1.060 | 0.011 | 1.028 | 1.006–1.051 |
| Stage | 0.000 | 2.012 | 1.667–2.427 | 0.000 | 1.696 | 1.400–2.055 |
| Grade | 0.001 | 2.696 | 1.777–4.088 | 0.065 | 1.522 | 0.975–2.735 |
| POLE mutation | 0.293 | 0.463 | 0.111–1.941 | |||
| P53 mutation | 0.329 | 1.411 | 0.707–2.817 | |||
| MSI | 0.331 | 1.158 | 0.862–1.565 | |||
| Risk score | 0.000 | 2.718 | 2.096–3.525 | 0.001 | 1.707 | 1.255–2.323 |
| Immune score | 0.023 | 1.001 | 1.000–1.002 | 0.166 | 1.001 | 1.000–1.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, T.; Wan, J.; Song, Q.; Chen, P.; Li, X. Identification of a Novel Epithelial–Mesenchymal Transition-Related Gene Signature for Endometrial Carcinoma Prognosis. Genes 2022, 13, 216. https://doi.org/10.3390/genes13020216
Ruan T, Wan J, Song Q, Chen P, Li X. Identification of a Novel Epithelial–Mesenchymal Transition-Related Gene Signature for Endometrial Carcinoma Prognosis. Genes. 2022; 13(2):216. https://doi.org/10.3390/genes13020216
Chicago/Turabian StyleRuan, Tianyuan, Jing Wan, Qian Song, Peigen Chen, and Xiaomao Li. 2022. "Identification of a Novel Epithelial–Mesenchymal Transition-Related Gene Signature for Endometrial Carcinoma Prognosis" Genes 13, no. 2: 216. https://doi.org/10.3390/genes13020216
APA StyleRuan, T., Wan, J., Song, Q., Chen, P., & Li, X. (2022). Identification of a Novel Epithelial–Mesenchymal Transition-Related Gene Signature for Endometrial Carcinoma Prognosis. Genes, 13(2), 216. https://doi.org/10.3390/genes13020216