
Evaluation of Bacteria and Fungi DNA Abundance in Human Tissues

 , ,
, ,  , , and add
Show full author list
, , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction Methods
2.2. Primer Pairs and qPCR DNA Amplification
2.3. Standard Curves
2.4. Shotgun Sequencing
2.5. Statistical Analysis
3. Results
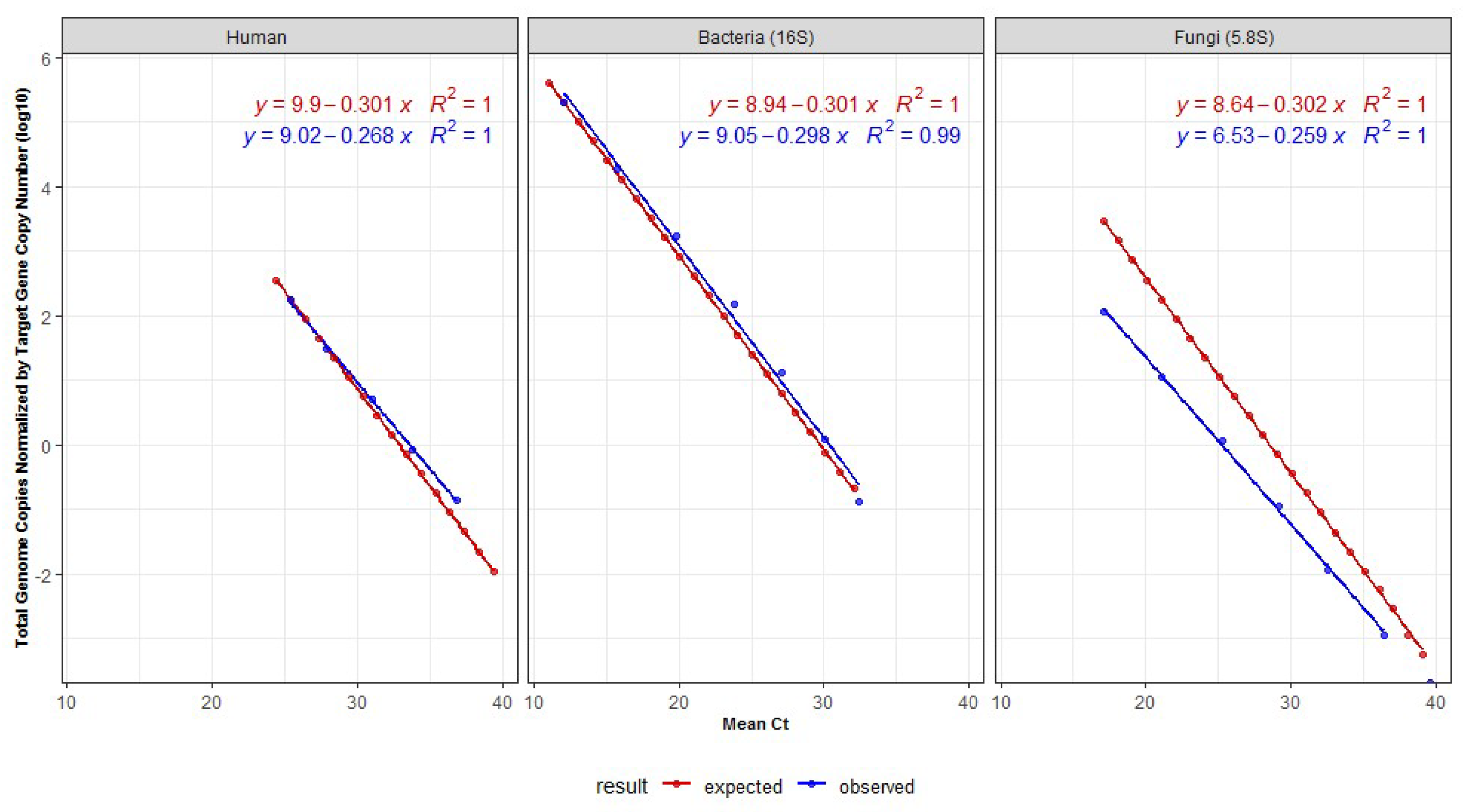
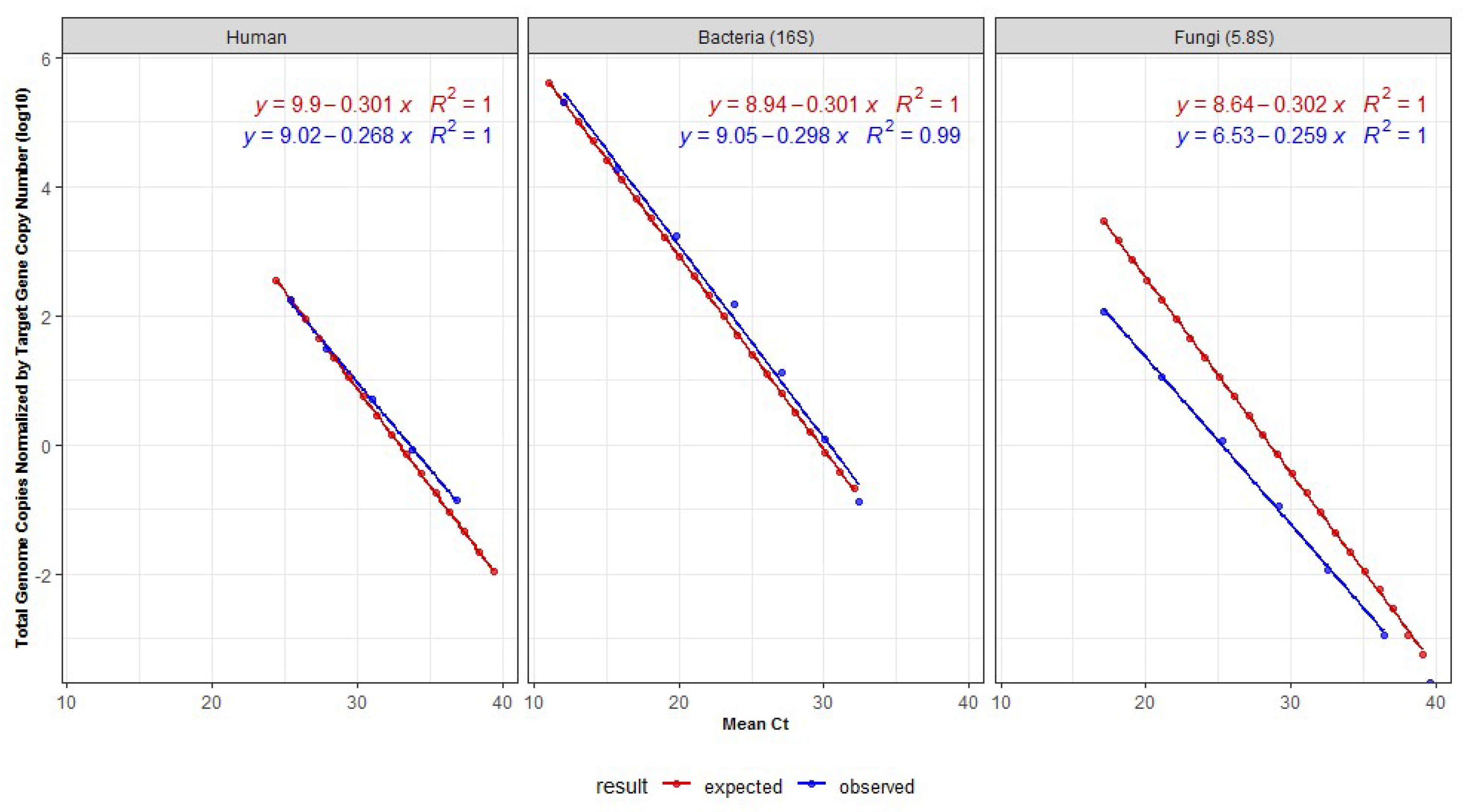
3.1. Standard Curves Correlating Genome Copies and Cts
3.2. Impacts of Varying Bacteria:Human and Fungi:Human DNA-Ratios over Amplification/Quantification Capabilities
3.3. Genome Copies in Different Sample-Types
3.4. Shotgun Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, Z.; Doty, S.L. Characterization of bacterial endophytes of sweet potato plants. Plant Soil 2009, 322, 197–207. [Google Scholar] [CrossRef]
- Hardoim, P.R.; van Overbeek, L.S.; van Elsas, J.D. Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 2008, 6, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P.; Kolarik, M.; Štursová, M.; Kopecky, J.; Valášková, V.; Větrovský, T.; Žifčáková, L.; Šnajdr, J.; Rídl, J.; Vlček, Č.; et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auffret, M.D.; Karhu, K.; Khachane, A.; Dungait, J.; Fraser, F.; Hopkins, D.; Wookey, P.; Singh, B.; Freitag, T.E.; Hartley, I.P.; et al. The Role of Microbial Community Composition in Controlling Soil Respiration Responses to Temperature. PLoS ONE 2016, 11, e0165448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, C.; Afshinnekoo, E.; Ahsannudin, S.; Ghedin, E.; Read, T.; Fraser, C.; Dudley, J.; Hernandez, M.; Bowler, C.; Stolovitzky, G.; et al. The Metagenomics and Metadesign of the Subways and Urban Biomes (MetaSUB) International Consortium inaugural meeting report. Microbiome 2016, 4, 24. [Google Scholar]
- Danko, D.; Bezdan, D.; Afshin, E.E.; Ahsanuddin, S.; Bhattacharya, C.; Butler, D.J.; Chng, K.R.; Donnellan, D.; Hecht, J.; Jackson, K.; et al. A global metagenomic map of urban microbiomes and antimicrobial resistance. Cell 2021, 184, 3376–3393.e17. [Google Scholar] [CrossRef]
- Lax, S.; Sangwan, N.; Smith, D.; Larsen, P.; Handley, K.M.; Richardson, M.; Guyton, K.; Krezalek, M.; Shogan, B.D.; Defazio, J.; et al. Bacterial colonization and succession in a newly opened hospital. Sci. Transl. Med. 2017, 9, eaah6500. [Google Scholar] [CrossRef] [Green Version]
- Chng, K.R.; Li, C.; Bertrand, D.; Ng, A.H.Q.; Kwah, J.S.; Low, H.M.; Tong, C.; Natrajan, M.; Zhang, M.H.; Xu, L.; et al. Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 2020, 26, 941–951. [Google Scholar] [CrossRef]
- Checinska Sielaff, A.; Urbaniak, C.; Mohan, G.B.M.; Stepanov, V.G.; Tran, Q.; Wood, J.M.; Minich, J.; McDonald, D.; Mayer, T.; Knight, R.; et al. Characterization of the total and viable bacterial and fungal communities associated with the International Space Station surfaces. Microbiome 2019, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Biasucci, G.; Rubini, M.; Riboni, S.; Morelli, L.; Bessi, E.; Retetangos, C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum. Dev. 2010, 86 (Suppl. 1), 13–15. [Google Scholar] [CrossRef]
- Mueller, N.T.; Whyatt, R.; Hoepner, L.; Oberfield, S.; Dominguez-Bello, M.G.; Widen, E.M.; Hassoun, A.; Perera, F.; Rundle, A. Prenatal exposure to antibiotics, cesarean section and risk of childhood obesity. Int. J. Obes. 2015, 39, 665–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S. Determining the Presence of Periodontopathic Virulence Factors in Short-Term Postmortem Alzheimer’s Disease Brain Tissue. J. Alzheimer’s Dis. 2013, 36, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Jesus, E.C.; Lopes, A.; Aguiar, S.J.; Begnami, M.D.; Rocha, R.M.; Carpinetti, P.A.; Camargo, A.A.; Hoffmann, C.; Freitas, H.C.; et al. Tissue-Associated Bacterial Alterations in Rectal Carcinoma Patients Revealed by 16S rRNA Community Profiling. Front. Cell. Infect. Microbiol. 2016, 6, 179. [Google Scholar] [CrossRef] [Green Version]
- Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation|Nature Medicine. Available online: https://www.nature.com/articles/s41591-019-0405-7 (accessed on 20 January 2022).
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 2018, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Heravi, F.S.; Zakrzewski, M.; Vickery, K.; Hu, H. Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples. J. Microbiol. Methods 2020, 170, 105856. [Google Scholar] [CrossRef]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; van Zanten, S.J.O.V. Differences between Tissue-Associated Intestinal Microfloras of Patients with Crohn’s Disease and Ulcerative Colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.M.; Gleber-Netto, F.O.; Fernandes, G.R.; Amorim, M.; Barbosa, L.F.; Francisco, A.L.N.; de Andrade, A.G.; Setubal, J.C.; Kowalski, L.P.; Nunes, D.N.; et al. Alcohol and tobacco consumption affects bacterial richness in oral cavity mucosa biofilms. BMC Microbiol. 2014, 14, 250. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; De Simone, G.; Gioia, R.; Noviello, S.; Pagliara, D.; Campitiello, N.; Rubino, C.; Pardo, D.L.; Boccia, G.; DE Caro, F.; et al. Deep tissue biopsy vs. superficial swab culture, including microbial loading determination, in the microbiological assessment of Skin and Soft Tissue Infections (SSTIs). J. Chemother. 2017, 29, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Donachie, S.P.; Foster, J.S.; Brown, M.V. Culture clash: Challenging the dogma of microbial diversity. ISME J. 2007, 1, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Campanaro, S.; Treu, L.; Kougias, P.G.; Zhu, X.; Angelidaki, I. Taxonomy of anaerobic digestion microbiome reveals biases associated with the applied high throughput sequencing strategies. Sci. Rep. 2018, 8, 1926. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, S.; Calin, G.A. Gut microbiota: A new player in regulating immune- and chemo-therapy efficacy. Cancer Drug Resist. 2020, 3, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Chng, K.R.; Chan, S.H.; Ng, A.H.Q.; Li, C.; Jusakul, A.; Bertrand, D.; Wilm, A.; Choo, S.P.; Tan, D.M.Y.; Lim, K.H.; et al. Tissue Microbiome Profiling Identifies an Enrichment of Specific Enteric Bacteria in Opisthorchis viverrini Associated Cholangiocarcinoma. eBioMedicine 2016, 8, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, J.; Park, S.-J. High-throughput 16S rRNA gene sequencing reveals alterations of mouse intestinal microbiota after radiotherapy. Anaerobe 2015, 33, 1–7. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Pérez, J.C. Fungi of the human gut microbiota: Roles and significance. Int. J. Med. Microbiol. 2021, 311, 151490. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Zhang, S.; Li, H.; Yang, F.; Mushtaq, N.; Ullah, S.; Shi, Y.; An, C.; Xu, J. The influence of gut microbiota dysbiosis to the efficacy of 5-Fluorouracil treatment on colorectal cancer. Biomed. Pharmacother. 2018, 108, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.; Gupta, S.; Daily, J.; Kelly, L. Human microbiome signatures of differential colorectal cancer drug metabolism. NPJ Biofilms Microbiomes 2017, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.T.; Forche, A.; Selmecki, A. Ploidy Variation in Fungi: Polyploidy, Aneuploidy, and Genome Evolution. Microbiol. Spectr. 2017, 5, 599–618. [Google Scholar] [CrossRef] [Green Version]
- Wertheimer, N.B.; Stone, N.; Berman, J. Ploidy dynamics and evolvability in fungi. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150461. [Google Scholar] [CrossRef] [Green Version]
- Fredricks, D.N.; Smith, C.; Meier, A. Comparison of Six DNA Extraction Methods for Recovery of Fungal DNA as Assessed by Quantitative PCR. J. Clin. Microbiol. 2005, 43, 5122–5128. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, M.F.; Marshall, J.C. Source control in the management of sepsis. Intensiv. Care Med. 2001, 27, S49–S62. [Google Scholar] [CrossRef]
- Wenzel, R.P. Surgical site infections and the microbiome: An updated perspective. Infect. Control Hosp. Epidemiol. 2019, 40, 590–596. [Google Scholar] [CrossRef]
- Subramaniam, N.; Muthukrishnan, A. Oral mucositis and microbial colonization in oral cancer patients undergoing radiotherapy and chemotherapy: A prospective analysis in a tertiary care dental hospital. J. Investig. Clin. Dent. 2019, 10, e12454. [Google Scholar] [CrossRef]
- Irfan, M.; Delgado, R.Z.R.; Frias-Lopez, J. The Oral Microbiome and Cancer. Front. Immunol. 2020, 11, 591088. [Google Scholar] [CrossRef]
- He, J.-Y.; Wang, W.-Z.; Qi, H.-Z.; Ma, Y.; He, S.-Y. Use of recombinant Lactobacillus sakei for the prevention and treatment of radiation-induced enteritis. Med. Hypotheses 2018, 119, 37–40. [Google Scholar] [CrossRef]
- Riehl, T.E.; Alvarado, D.; Ee, X.; Zuckerman, A.; Foster, L.; Kapoor, V.; Thotala, D.; Ciorba, M.A.; Stenson, W.F. Lactobacillus rhamnosus GG protects the intestinal epithelium from radiation injury through release of lipoteichoic acid, macrophage activation and the migration of mesenchymal stem cells. Gut 2018, 68, 1003–1013. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism Group | DNA Mass in qPCR (ng) | Mean Ct | Genome Lenght (Mb) | Number of Genome Copies | Number of Genomes Normalized by Copies of the Target | Log10 of the Number Genomes |
|---|---|---|---|---|---|---|
| Mock-bacteria | 4.800000 | 12.1 | 3.6 | 1,242,233.8 | 207,038.9 | 5.3 |
| 0.432432 | 15.8 | 3.6 | 111,913.0 | 18,652.16 | 4.3 | |
| 0.038958 | 19.8 | 3.6 | 10,082.2 | 1680.37 | 3.2 | |
| 0.003510 | 23.8 | 3.6 | 908.3 | 151.39 | 2.2 | |
| 0.000316 | 27.1 | 3.6 | 81.8 | 13.64 | 1.1 | |
| 0.000028 | 30.0 | 3.6 | 7.4 | 1.23 | 0.1 | |
| 0.000003 | 32.4 | 3.6 | 0.7 | 0.11 | −1.0 | |
| Human | 5.000000 | 25.4 | 64,000 | 723.9 | 180.96 | 2.3 |
| 0.833333 | 27.8 | 64,000 | 120.6 | 30.16 | 1.5 | |
| 0.138889 | 31.1 | 64,000 | 20.1 | 5.03 | 0.7 | |
| 0.023148 | 33.8 | 64,000 | 3.4 | 0.84 | −0.1 | |
| 0.003858 | 36.9 | 64,000 | 0.6 | 0.14 | −0.9 | |
| Mock-fungi | 0.200000 | 17.1 | 15.6 | 11,875.1 | 115.9 | 2.1 |
| 0.020000 | 21.1 | 15.6 | 1187.5 | 11.6 | 1.1 | |
| 0.002000 | 25.3 | 15.6 | 118.8 | 1.2 | 0.06 | |
| 0.000200 | 29.2 | 15.6 | 11.9 | 0.1 | −0.9 | |
| 0.000020 | 32.6 | 15.6 | 1.2 | 0.01 | −1.9 | |
| 0.000002 | 36.4 | 15.6 | 0.1 | 0.001 | −2.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Albuquerque, G.E.; Moda, B.S.; Serpa, M.S.; Branco, G.P.; Defelicibus, A.; Takenaka, I.K.T.M.; de Amorim, M.G.; Miola, E.C.; Martins, V.C.A.; Torres, K.L.; et al. Evaluation of Bacteria and Fungi DNA Abundance in Human Tissues. Genes 2022, 13, 237. https://doi.org/10.3390/genes13020237
de Albuquerque GE, Moda BS, Serpa MS, Branco GP, Defelicibus A, Takenaka IKTM, de Amorim MG, Miola EC, Martins VCA, Torres KL, et al. Evaluation of Bacteria and Fungi DNA Abundance in Human Tissues. Genes. 2022; 13(2):237. https://doi.org/10.3390/genes13020237
Chicago/Turabian Stylede Albuquerque, Gabriela E., Bruno S. Moda, Marianna S. Serpa, Gabriela P. Branco, Alexandre Defelicibus, Isabella K. T. M. Takenaka, Maria G. de Amorim, Elizabeth C. Miola, Valquiria C. A. Martins, Katia L. Torres, and et al. 2022. "Evaluation of Bacteria and Fungi DNA Abundance in Human Tissues" Genes 13, no. 2: 237. https://doi.org/10.3390/genes13020237
APA Stylede Albuquerque, G. E., Moda, B. S., Serpa, M. S., Branco, G. P., Defelicibus, A., Takenaka, I. K. T. M., de Amorim, M. G., Miola, E. C., Martins, V. C. A., Torres, K. L., Bezerra, S. M., Claro, L. C. L., Pelosof, A. G., Sztokfisz, C. Z., Abrantes, L. L. S., Coimbra, F. J. F., Kowalski, L. P., Alves, F. A., Zequi, S. C., ... Dias-Neto, E. (2022). Evaluation of Bacteria and Fungi DNA Abundance in Human Tissues. Genes, 13(2), 237. https://doi.org/10.3390/genes13020237