
Mucolipidosis Type IV in Omani Families with a Novel MCOLN1 Mutation: Search for Evidence of Founder Effect

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Enrolment and Clinical Characterization
2.2. Confirmation of MCOLN1 Mutation by DNA SANGER Sequencing
2.3. Mutation Screening in the General Population
3. Results
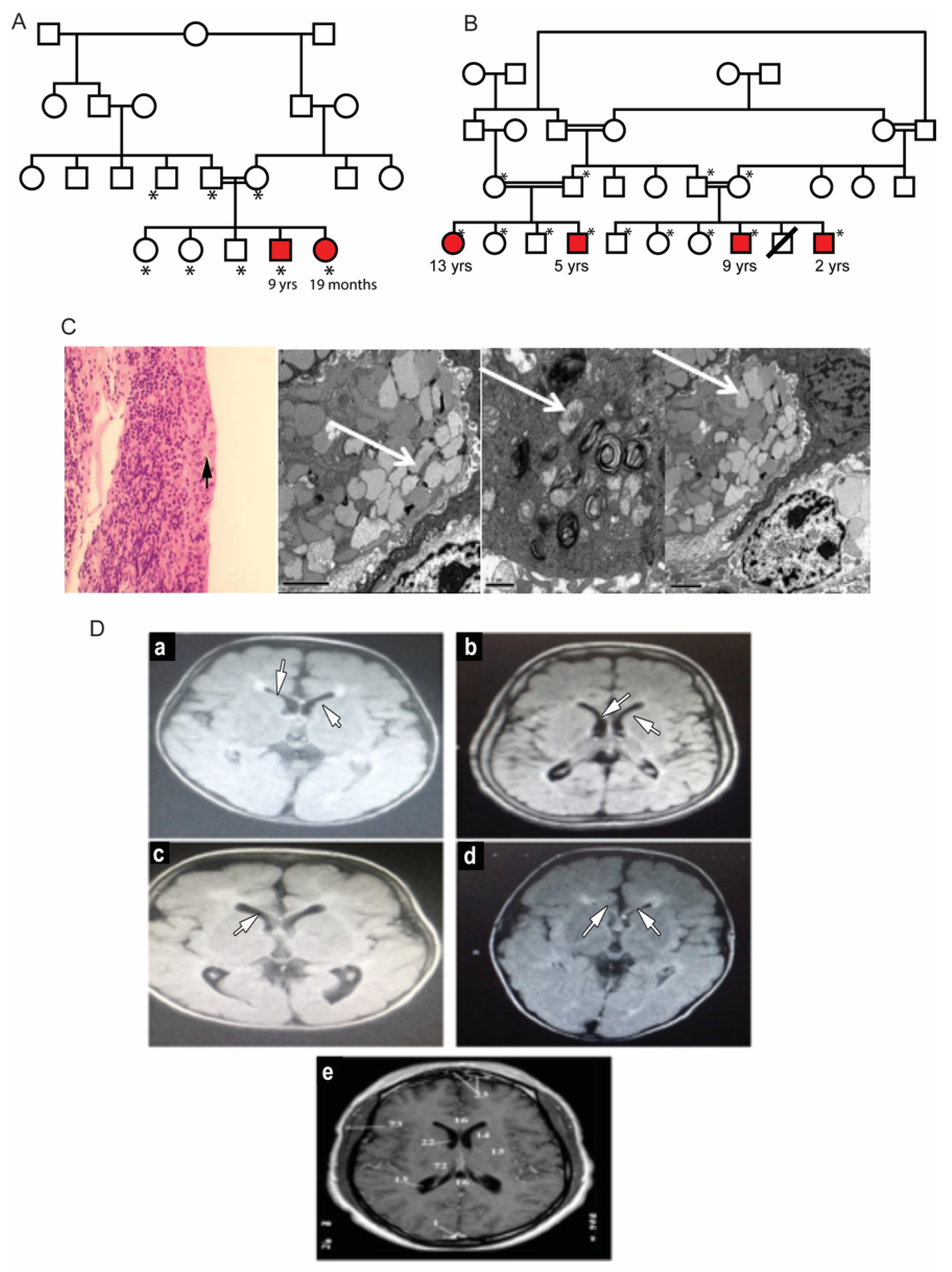
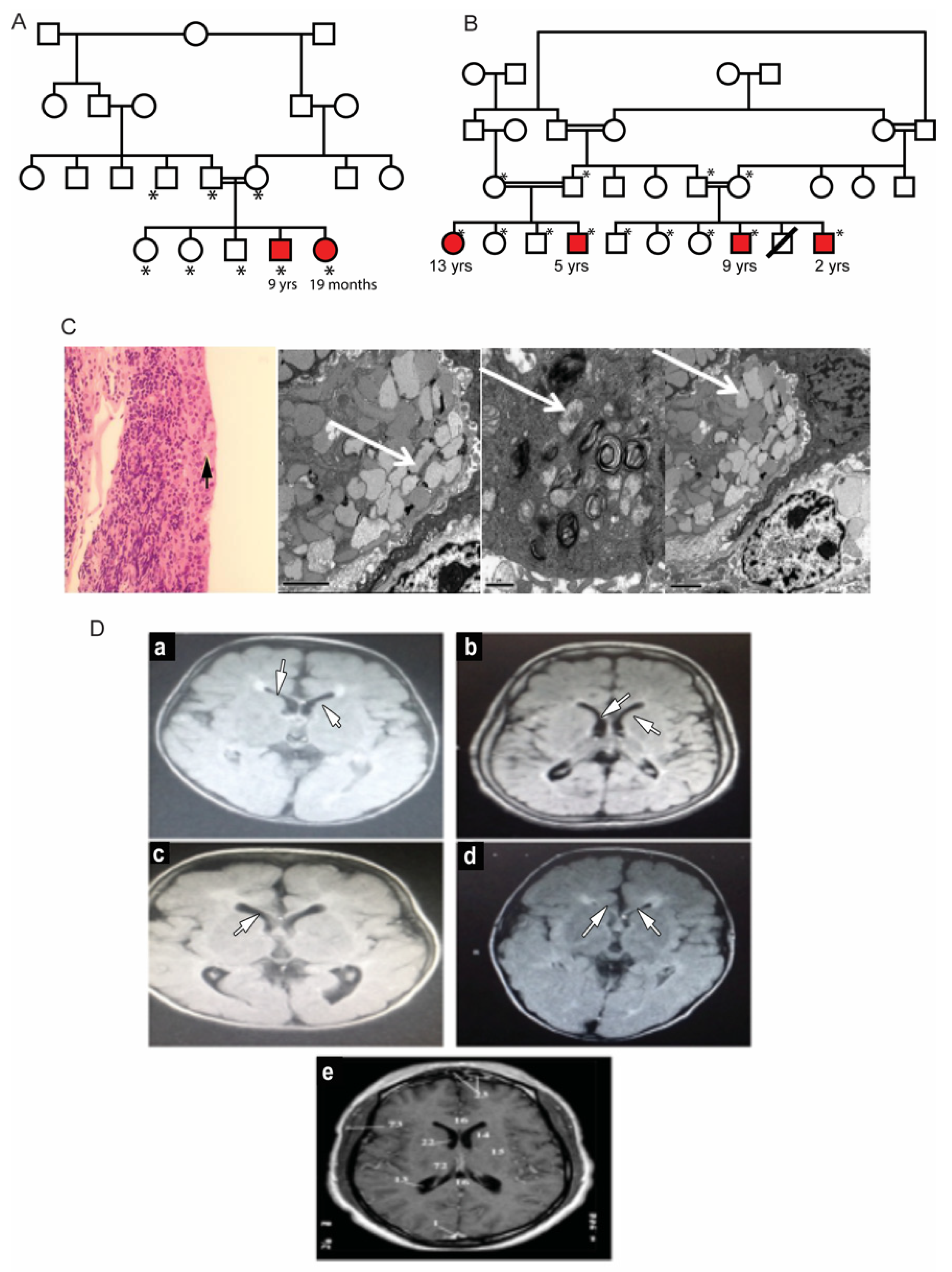
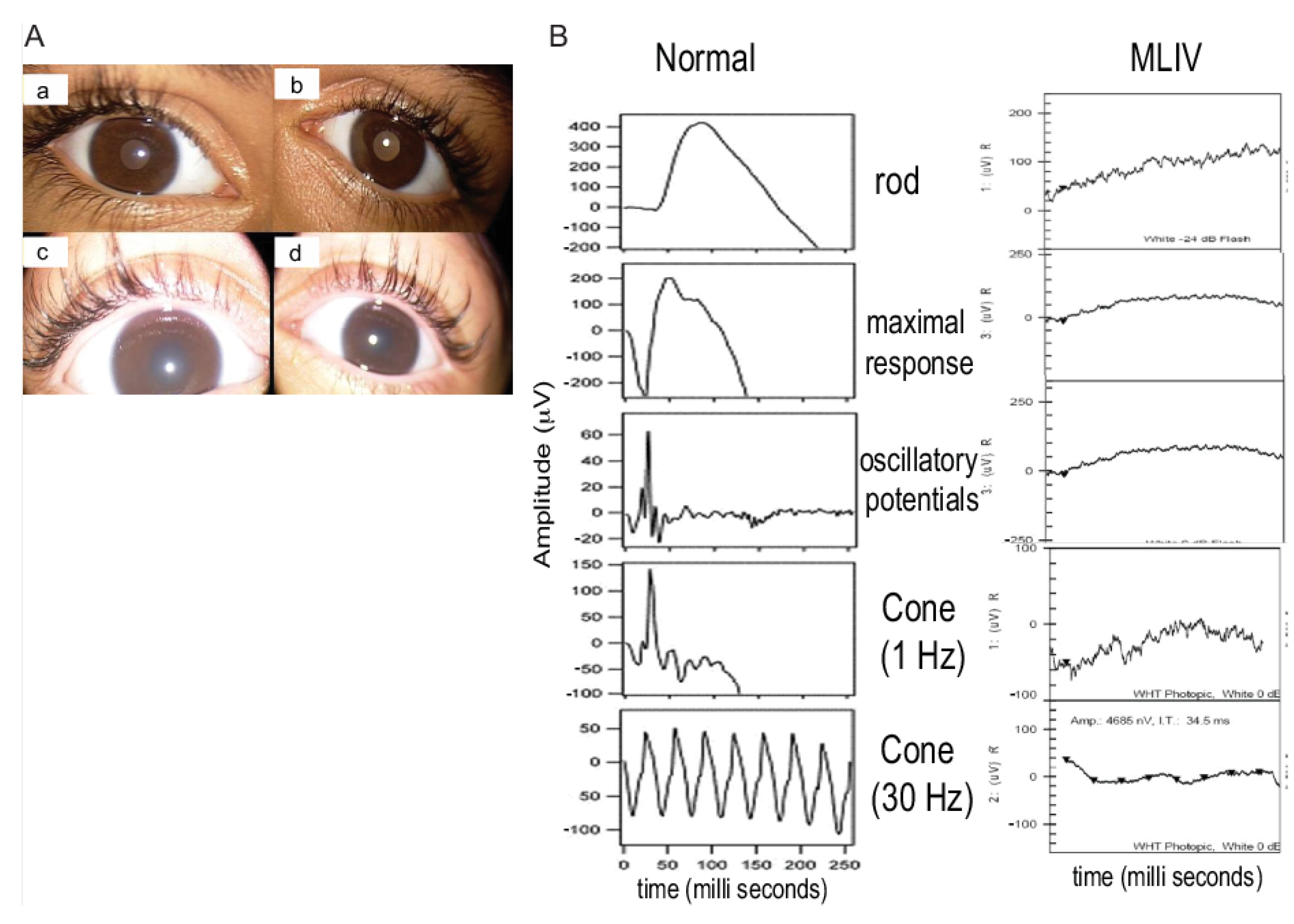
3.1. Clinical Description of Patients in Two Families
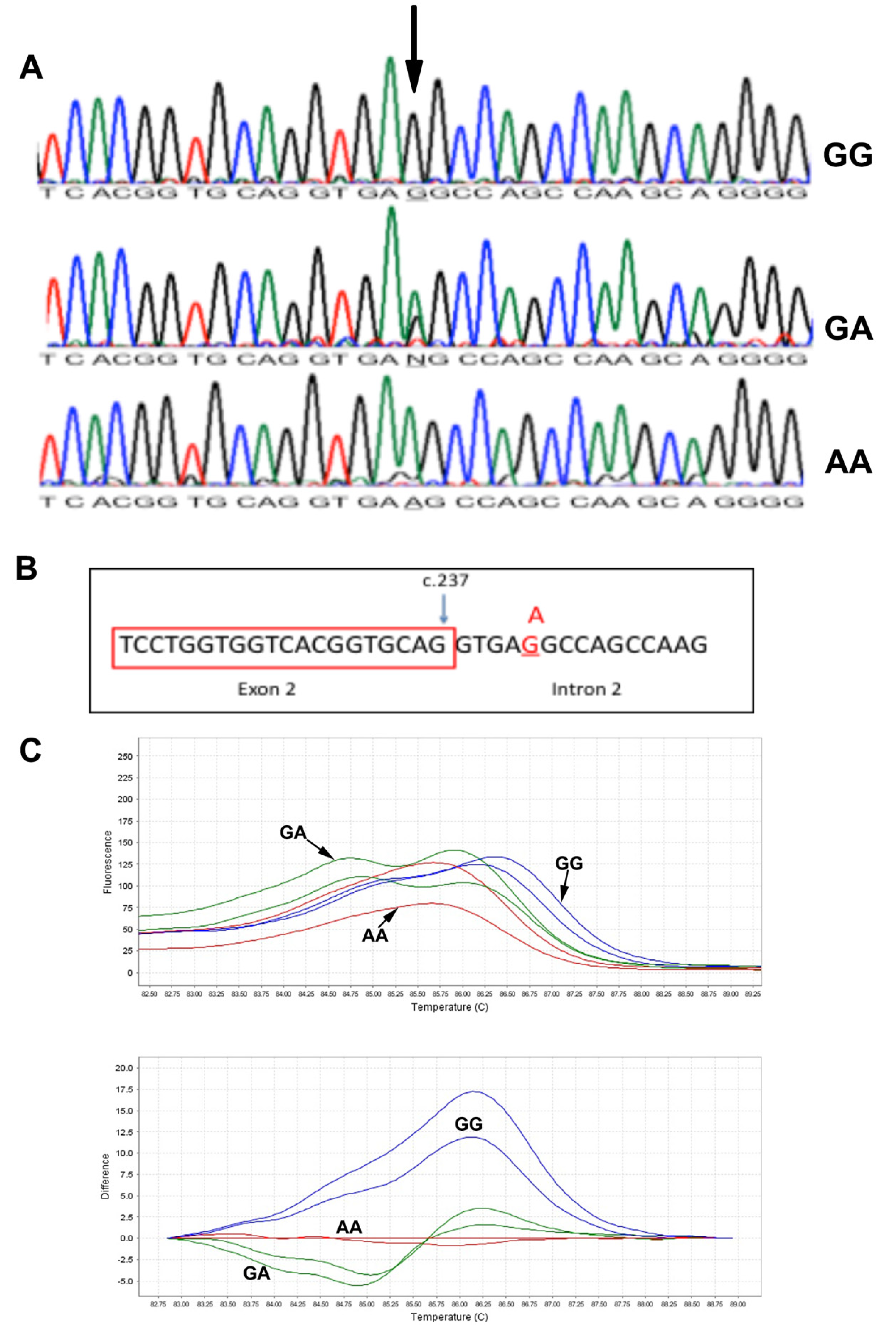
3.2. Mutation Analysis of MCOLN1 Gene and Population Screening
3.3. c.237+5 G>A Analysis with High-Resolution Melting and Determination of Founder Effect
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LaPlante, J.M.; Sun, M.; Falardeau, J.; Dai, D.; Brown, E.M.; Slaugenhaupt, S.A.; Vassilev, P.M. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol. Genet. Metab. 2006, 89, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Gustafson, A.M.; Sidransky, E.; Goldin, E. Mucolipidosis type IV: An update. Mol. Genet. Metab. 2011, 104, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Thihli, K.; Al-Murshedi, F.; Al-Hashmi, N.; Al-Mamari, W.; Islam, M.M.; Al-Yahyaee, S.A. Consanguinity, endogamy and inborn errors of metabolism in Oman: A cross-sectional study. Hum. Hered. 2014, 77, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.N.; Hashim, J.; Venugopalan, P. Pattern of inborn errors of metabolism in an Omani population of the Arabian Peninsula. Ann. Trop. Paediatr. 2002, 22, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.O.; Jaju, D.; Albarwani, S.; Al-Yahyaee, S.; Al-Hadabi, S.; Lopez-Alvarenga, J.C.; Rizvi, S.G.; Comuzzie, A.G.; Bayoumi, R.A. Non-dipping blood pressure in the metabolic syndrome among Arabs of the Oman family study. Obesity. 2007, 15, 2445–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadjali, F.; Al-Yahyaee, S.; Hassan, M.O.; Albarwani, S.; Bayoumi, R.A. Association of adiponectin promoter variants with traits and clusters of metabolic syndrome in Arabs: Family-based study. Gene 2013, 527, 663–669. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Bindu, P.S.; Gayathri, N.; Yasha, T.C.; Kovoor, J.M.; Subasree, R.; Rao, S.; Panda, S.; Pal, P.K. A variant form of mucolipidosis IV: Report on 4 patients from the Indian subcontinent. J. Child Neurol. 2008, 23, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli-Badenier, M.; Severino, M.; Tappino, B.; Tortora, D.; Camia, F.; Zanaboni, C.; Brera, F.; Priolo, E.; Rossi, A.; Biancheri, R.; et al. A novel homozygous MCOLN1 double mutant allele leading to TRP channel domain ablation underlies Mucolipidosis IV in an Italian Child. Metab. Brain Dis. 2015, 30, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Tuysuz, B.; Goldin, E.; Metin, B.; Korkmaz, B.; Yalcinkaya, C. Mucolipidosis type IV in a Turkish boy associated with a novel MCOLN1 mutation. Brain Dev. 2009, 31, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Reis, S.; Sheffer, R.N.; Merin, S.; Luder, A.S.; Bach, G. Mucolipidosis type IV: A mild form with late onset. Am. J. Med. Genet. 1993, 47, 392–394. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Family 1 | Family 2 | |||||
|---|---|---|---|---|---|---|
| Patient # | 1 | 2 | 3 | 4 | 5 | 6 |
| Age | 9 yrs | 19 months | 13 yrs | 5 yrs | 9 yrs | 2 yrs |
| infantile onset of global D.D | + | + | + | + | + | + |
| Spastic quadriplegia | + | + | + | + | + | + |
| Contractures | + | − | − | − | + | + |
| Microcephaly | + | − | + | + | + | + |
| Dysmorphism | + | + | + | + | + | + |
| Blood Biochemistry: | ||||||
| Iron (11–28 μmol/L) | 2 | 5 | 2 | 4 | 2 | ND |
| Ferritin (24–336 ng/mL) | 2 | 5 | 2 | 5 | 2 | ND |
| Hemoglobin (11–15 g/dL) | 6.1 | 104 | 7 | 9 | 9.4 | 6.9 |
| Gastrin (15–110 ng/L) | 706 | 1691 | 1474 | ND | 801 | ND |
| Ophthalmological findings: | ||||||
| Photophobia | − | − | + | − | + | + |
| Nystagmus | + | + | − | − | − | − |
| Strabismus | RX | RE | AXT | RE | LX | LX |
| Corneal haziness | + | ++ | +++ | +++ | +++ | +++ (Rt) ++ (Lt) |
| Pupil reaction to light | sluggish | sluggish | N | sluggish | N | sluggish |
| Pigmentary retinopathy | + | + | No view | + | No view | No view |
| ERG- rod cone dysfunction | +++ | ND | +++ | +++ | +++ | +++ |
| Brian MRI: | ||||||
| Thin corpus callosum | + | + | + | + | + | + |
| Periventricular white matter changes | + | + | + | + | + | + |
| Splice Site | Sequence | MAXENT | MDD | MM | WMM |
|---|---|---|---|---|---|
| wild-type | cag*GTGAGG | 10.07 | 13.38 | 10.18 | 10.76 |
| Patient | cag*GTGAAG | 6.66 | 11.18 | 7.41 | 7.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Alawi, B.; Harikrishna, B.; Al-Thihli, K.; Al Zuhabi, S.; Ganesh, A.; Al Hashami, Z.; Al Dhamhmani, Z.; Zadjali, R.; Al Riyami, N.B.; Zadjali, F. Mucolipidosis Type IV in Omani Families with a Novel MCOLN1 Mutation: Search for Evidence of Founder Effect. Genes 2022, 13, 248. https://doi.org/10.3390/genes13020248
Al-Alawi B, Harikrishna B, Al-Thihli K, Al Zuhabi S, Ganesh A, Al Hashami Z, Al Dhamhmani Z, Zadjali R, Al Riyami NB, Zadjali F. Mucolipidosis Type IV in Omani Families with a Novel MCOLN1 Mutation: Search for Evidence of Founder Effect. Genes. 2022; 13(2):248. https://doi.org/10.3390/genes13020248
Chicago/Turabian StyleAl-Alawi, Badriya, Beena Harikrishna, Khalid Al-Thihli, Sana Al Zuhabi, Anuradha Ganesh, Zainab Al Hashami, Zeyana Al Dhamhmani, Razan Zadjali, Nafila B. Al Riyami, and Fahad Zadjali. 2022. "Mucolipidosis Type IV in Omani Families with a Novel MCOLN1 Mutation: Search for Evidence of Founder Effect" Genes 13, no. 2: 248. https://doi.org/10.3390/genes13020248
APA StyleAl-Alawi, B., Harikrishna, B., Al-Thihli, K., Al Zuhabi, S., Ganesh, A., Al Hashami, Z., Al Dhamhmani, Z., Zadjali, R., Al Riyami, N. B., & Zadjali, F. (2022). Mucolipidosis Type IV in Omani Families with a Novel MCOLN1 Mutation: Search for Evidence of Founder Effect. Genes, 13(2), 248. https://doi.org/10.3390/genes13020248