
Proteogenomics Integrating Reveal a Complex Network, Alternative Splicing, Hub Genes Regulating Heart Maturation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Heart Tissue Preparation
2.2. Protein Extraction, Peptide Digestion, and Analysis by LC-MS/MS
2.3. Protein Identification and Quantification Based on iTRAQ Data
2.4. RNA Extraction, Library Preparation, and Transcriptome Sequencing
2.5. Processing of Sequence Data and Mapping Reads to Reference Genome
2.6. Functional Enrichment Analysis of Differentially Expressed Proteins and Differentially Expressed Genes
2.7. Alternative Splicing Genes Analysis
2.8. Hub Genes Analysis
2.9. Quantitative Real-Time PCR Analysis
2.10. Primary Cardiomyocytes Mitochondrial Stress Assay
3. Results
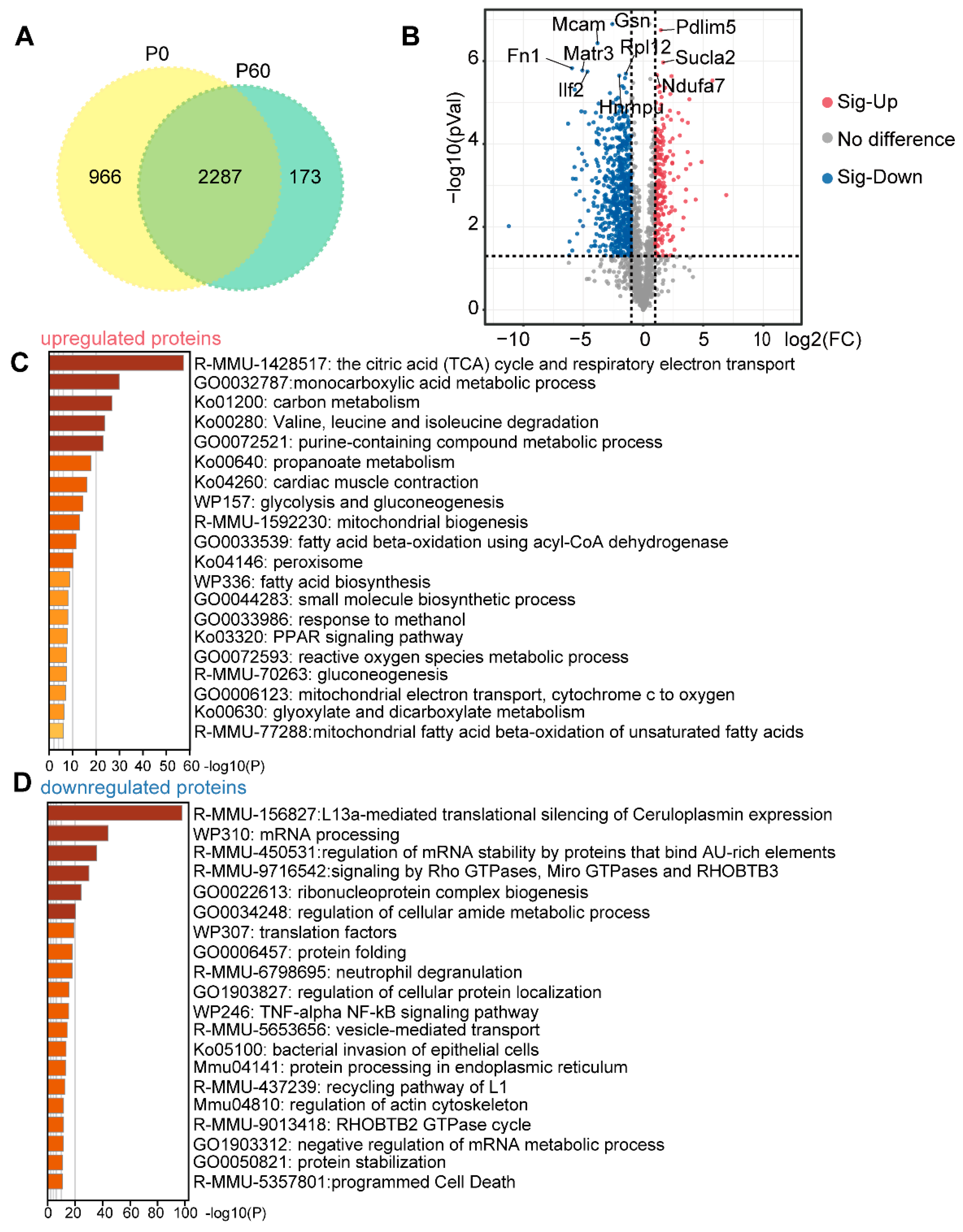
3.1. Proteomic Profiling of Neonatal and Adult Heart
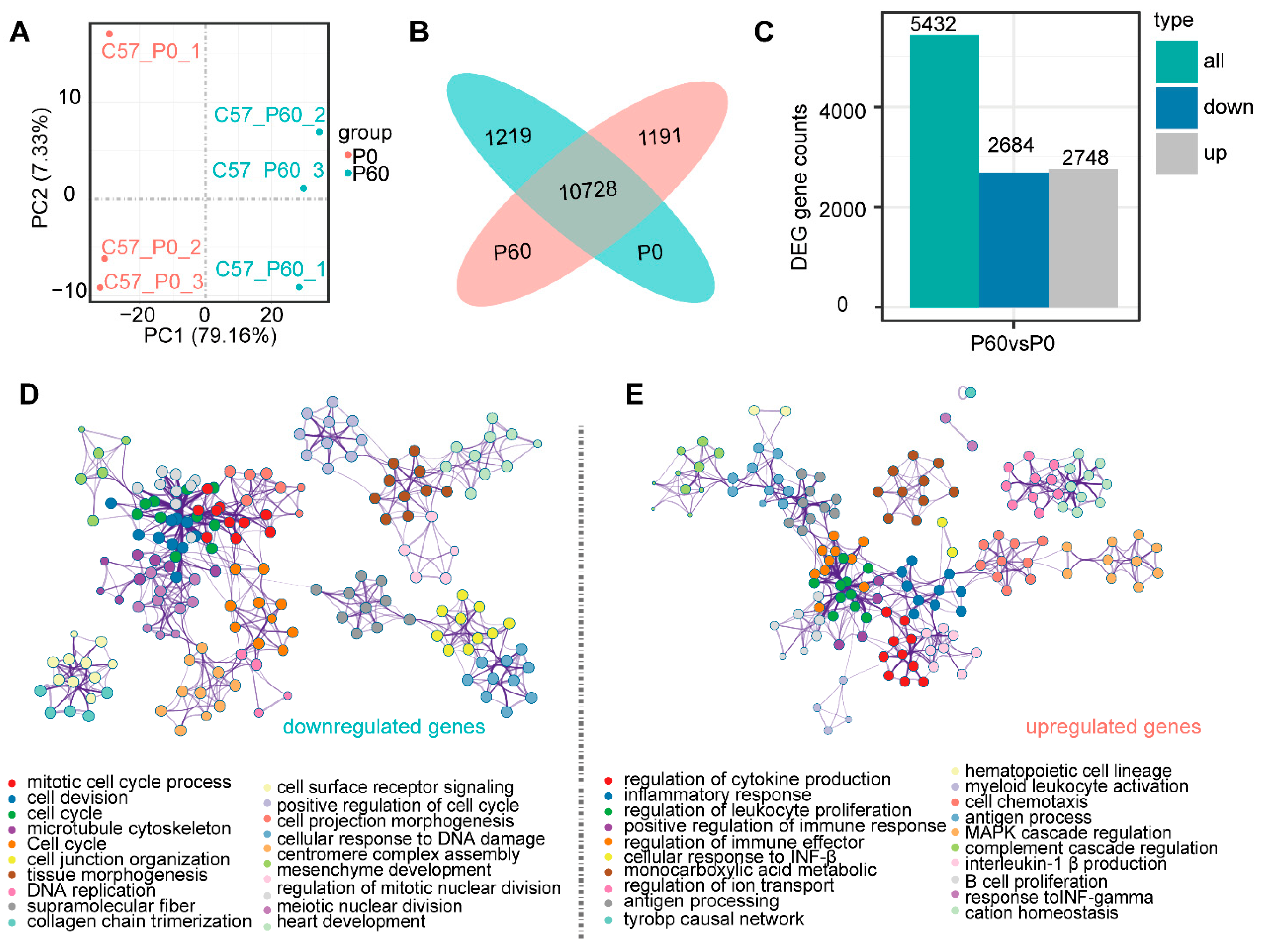
3.2. Transcriptomic Profiling of Neonatal and Adult Heart
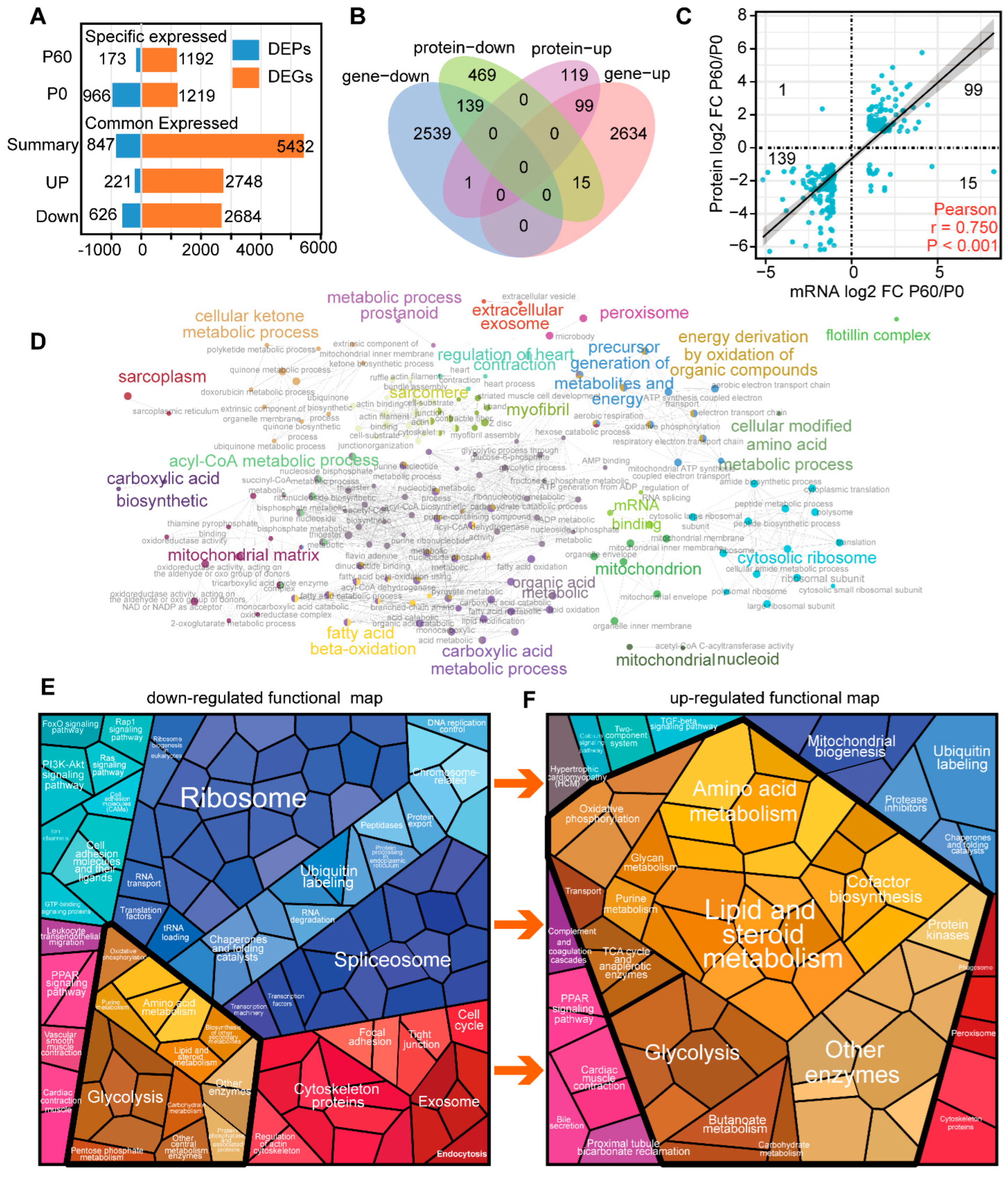
3.3. Compare Neonatal and Adult Heart by Proteomic and Transcriptome Combined Analysis
3.4. Alternative Splicing Mediating mRNA Process in Neonatal and Adult Heart
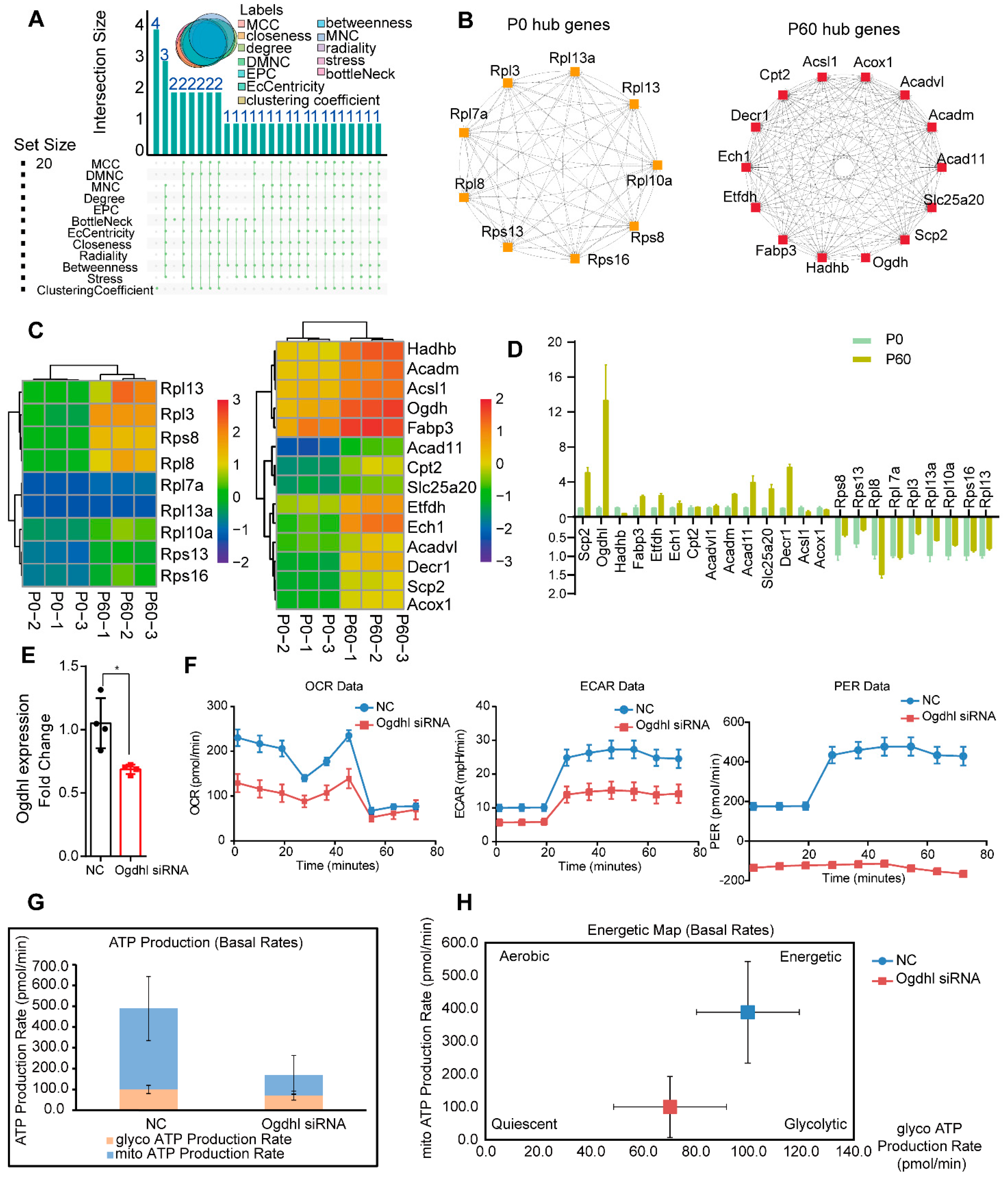
3.5. Network Analysis for hub Genes in Neonatal and Adult Heart
3.6. The Ogdhl Gene Play Essential Role in Cardiomyocyte Energy Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zaffran, S.; Frasch, M. Early signals in cardiac development. Circ. Res. 2002, 91, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Guo, Y.; Cao, Y.; Jardin, B.D.; Sethi, I.; Ma, Q.; Moghadaszadeh, B.; Troiano, E.C.; Mazumdar, N.; Trembley, M.A.; Small, E.M.; et al. Sarcomeres regulate murine cardiomyocyte maturation through MRTF-SRF signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2008861118. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Gunawan, F.; Ramadass, R.; Beisaw, A.; Konzer, A.; Mullapudi, S.T.; Gentile, A.; Maischein, H.M.; Graumann, J.; Stainier, D.Y.R. Mechanical Forces Regulate Cardiomyocyte Myofilament Maturation via the VCL-SSH1-CFL Axis. Dev. Cell 2019, 51, 62–77.e5. [Google Scholar] [CrossRef]
- Kannan, S.; Kwon, C. Regulation of cardiomyocyte maturation during critical perinatal window. J. Physiol. 2020, 598, 2941–2956. [Google Scholar] [CrossRef] [Green Version]
- Barton, P.J.; Bhavsar, P.K.; Brand, N.J.; Chan-Thomas, P.S.; Dabhade, N.; Farza, H.; Townsend, P.J.; Yacoub, M.H. Gene expression during cardiac development. Symp. Soc. Exp. Biol. 1992, 46, 251–264. [Google Scholar]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef] [Green Version]
- Pervolaraki, E.; Dachtler, J.; Anderson, R.A.; Holden, A.V. The developmental transcriptome of the human heart. Sci. Rep. 2018, 8, 15362. [Google Scholar] [CrossRef]
- Uosaki, H.; Cahan, P.; Lee, D.I.; Wang, S.; Miyamoto, M.; Fernandez, L.; Kass, D.A.; Kwon, C. Transcriptional Landscape of Cardiomyocyte Maturation. Cell Rep. 2015, 13, 1705–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Gunawan, F.; Beisaw, A.; Jimenez-Amilburu, V.; Maischein, H.M.; Kostin, S.; Kawakami, K.; Stainier, D.Y. Proteolysis regulates cardiomyocyte maturation and tissue integration. Nat. Commun. 2017, 8, 14495. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.; Qiu, X.T.; Sun, Q.; Zhou, J.P.; Yang, H.J.; Wu, W.Z.; He, L.F.; Tang, C.E.; Zhang, G.G.; Bai, Y.P. Endogenous reduction of miR-185 accelerates cardiac function recovery in mice following myocardial infarction via targeting of cathepsin K. J. Cell. Mol. Med. 2019, 23, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [Green Version]
- Otto, A.; Bernhardt, J.; Meyer, H.; Schaffer, M.; Herbst, F.A.; Siebourg, J.; Mäder, U.; Lalk, M.; Hecker, M.; Becher, D. Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nat. Commun. 2010, 1, 137. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, Y.; Hao, H.; Wang, Y.; Zhou, Z.; Wang, Z.; Chu, X. Screening Hub Genes as Prognostic Biomarkers of Hepatocellular Carcinoma by Bioinformatics Analysis. Cell Transplant. 2019, 28, 76s–86s. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.P.; Zhang, J.X.; Sun, Q.; Zhou, J.P.; Luo, J.M.; He, L.F.; Lin, X.C.; Zhu, L.P.; Wu, W.Z.; Wang, Z.Y.; et al. Induction of microRNA-199 by Nitric Oxide in Endothelial Cells Is Required for Nitrovasodilator Resistance via Targeting of Prostaglandin I2 Synthase. Circulation 2018, 138, 397–411. [Google Scholar] [CrossRef]
- Wang, J.; Chen, X.; Shen, D.; Ge, D.; Chen, J.; Pei, J.; Li, Y.; Yue, Z.; Feng, J.; Chu, M.; et al. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell. Cardiol. 2019, 127, 105–114. [Google Scholar] [CrossRef]
- Ai, S.; Peng, Y.; Li, C.; Gu, F.; Yu, X.; Yue, Y.; Ma, Q.; Chen, J.; Lin, Z.; Zhou, P.; et al. EED orchestration of heart maturation through interaction with HDACs is H3K27me3-independent. eLife 2017, 6, e24570. [Google Scholar] [CrossRef] [Green Version]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, J.; Song, S.; Li, H.; Yang, H.; Zhou, B.; Li, Y.; Yue, Z.; Lian, H.; Liu, L.; et al. Gp130 Controls Cardiomyocyte Proliferation and Heart Regeneration. Circulation 2020, 142, 967–982. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.T.; Ye, S.; Su, J.; Garg, V. Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration. Front. Cell Dev. Biol. 2020, 8, 594226. [Google Scholar] [CrossRef]
- Bertero, A.; Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C.E. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 2019, 10, 1538. [Google Scholar] [CrossRef] [Green Version]
- Giudice, J.; Xia, Z.; Wang, E.T.; Scavuzzo, M.A.; Ward, A.J.; Kalsotra, A.; Wang, W.; Wehrens, X.H.; Burge, C.B.; Li, W.; et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 2014, 5, 3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chen, Y.; Li, X.; Chen, G.; Zhong, L.; Chen, G.; Liao, Y.; Liao, W.; Bin, J. Genome-wide analysis of alternative splicing during human heart development. Sci. Rep. 2016, 6, 35520. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Lee, S.H.; Jung, Y.; Lee, Y.; Yoon, J.H.; Choi, J.Y.; Hwang, C.Y.; Son, Y.H.; Park, S.S.; Hwang, G.S.; et al. FABP3-mediated membrane lipid saturation alters fluidity and induces ER stress in skeletal muscle with aging. Nat. Commun. 2020, 11, 5661. [Google Scholar] [CrossRef]
- Violante, S.; Ijlst, L.; Te Brinke, H.; Tavares de Almeida, I.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Carnitine palmitoyltransferase 2 and carnitine/acylcarnitine translocase are involved in the mitochondrial synthesis and export of acylcarnitines. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 2039–2044. [Google Scholar] [CrossRef]
- Leslie, N.D.; Valencia, C.A.; Strauss, A.W.; Zhang, K. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. In GeneReviews (®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhou, Z.; Zhou, J.; Du, Y. Estrogen receptor beta interacts and colocalizes with HADHB in mitochondria. Biochem. Biophys. Res. Commun. 2012, 427, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Xu, L.; Yu, X.; Zhang, G.; Guo, H.; Liu, H.; Song, G.; Weng, S.; Dong, L.; Zhu, J.; et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J. Hepatol. 2020, 72, 909–923. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Liu, W.; Song, S.; Bai, L.; Nie, Y.; Bai, Y.; Zhang, G. Proteogenomics Integrating Reveal a Complex Network, Alternative Splicing, Hub Genes Regulating Heart Maturation. Genes 2022, 13, 250. https://doi.org/10.3390/genes13020250
Yang H, Liu W, Song S, Bai L, Nie Y, Bai Y, Zhang G. Proteogenomics Integrating Reveal a Complex Network, Alternative Splicing, Hub Genes Regulating Heart Maturation. Genes. 2022; 13(2):250. https://doi.org/10.3390/genes13020250
Chicago/Turabian StyleYang, Huijun, Weijing Liu, Shen Song, Lina Bai, Yu Nie, Yongping Bai, and Guogang Zhang. 2022. "Proteogenomics Integrating Reveal a Complex Network, Alternative Splicing, Hub Genes Regulating Heart Maturation" Genes 13, no. 2: 250. https://doi.org/10.3390/genes13020250
APA StyleYang, H., Liu, W., Song, S., Bai, L., Nie, Y., Bai, Y., & Zhang, G. (2022). Proteogenomics Integrating Reveal a Complex Network, Alternative Splicing, Hub Genes Regulating Heart Maturation. Genes, 13(2), 250. https://doi.org/10.3390/genes13020250