
Actionable Genomics in Clinical Practice: Paradigmatic Case Reports of Clinical and Therapeutic Strategies Based upon Genetic Testing


Abstract
:1. Introduction
2. Actionable Genomics in Psychiatry: Example of a 16p11.2 Duplication Case Report
Case Report
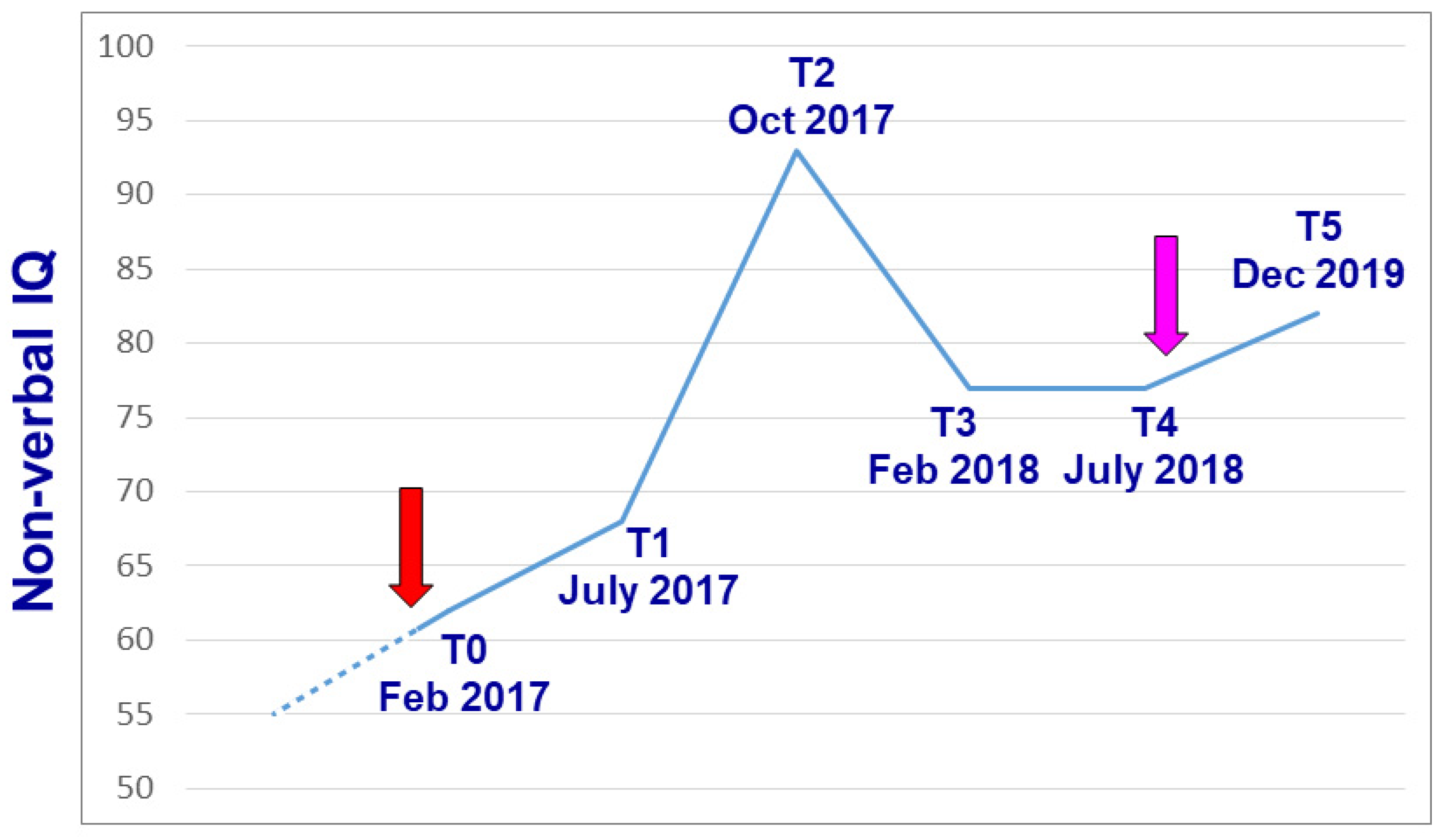
3. Genetically-Driven Psychopharmacotherapy: A Paradigmatic Case of Intellectual Disability and Autism Spectrum Disorder
Case Report
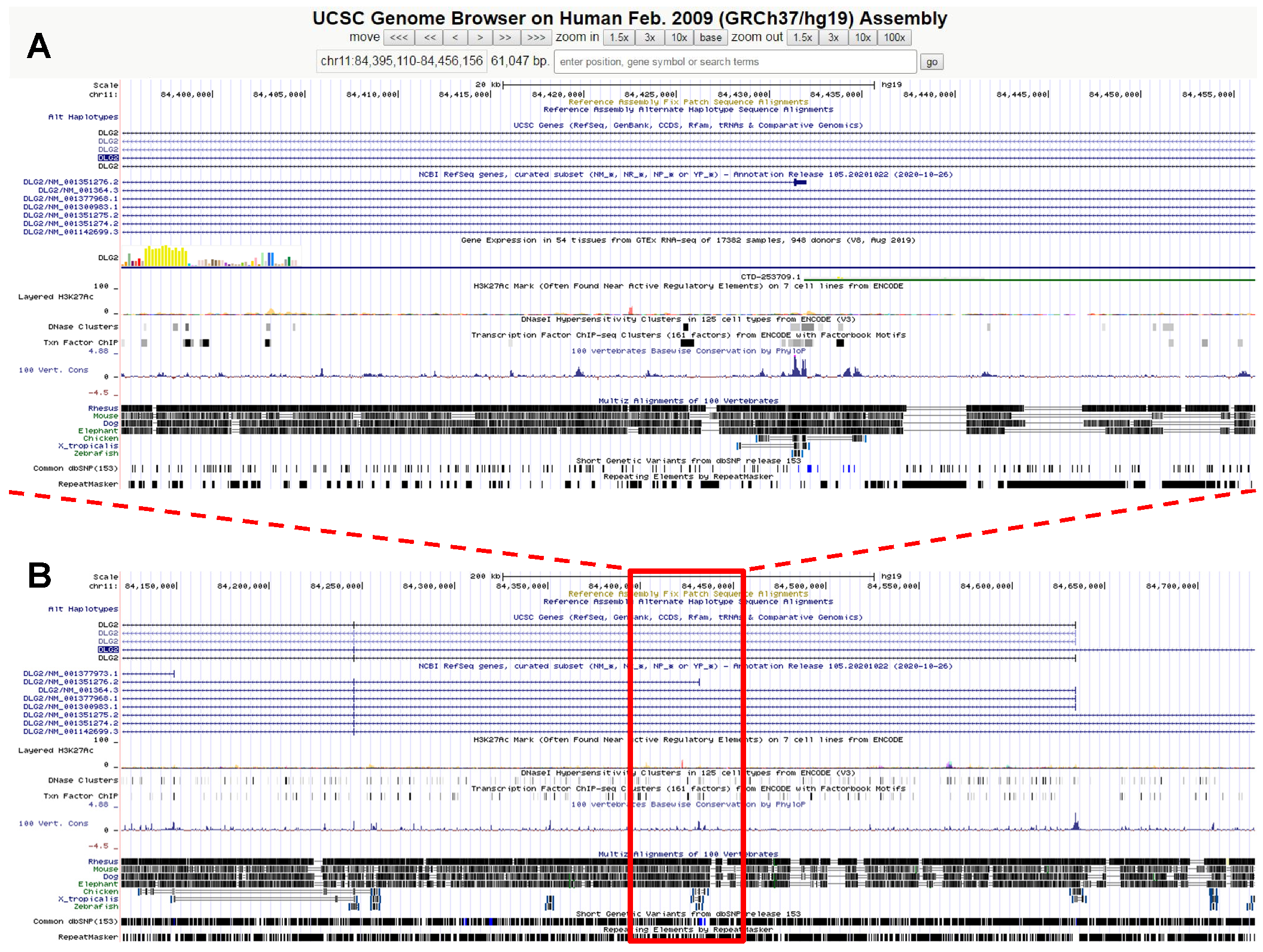
4. COL3A1 Gene Variant and Rupture of the Colon in an Extended Family with a Connective Tissue Disorder
Case Report
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leblond, C.S.; Le, T.L.; Malesys, S.; Cliquet, F.; Tabet, A.C.; Delorme, R.; Rolland, T.; Bourgeron, T. Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Mol. Cell. Neurosci. 2021, 113, 103623. [Google Scholar] [CrossRef]
- Persico, A.M.; Arango, C.; Buitelaar, J.K.; Correll, C.U.; Glennon, J.C.; Hoekstra, P.J.; Moreno, C.; Vitiello, B.; Vorstman, J.; The European Child and Adolescent Clinical Psychopharmacology Network; et al. Unmet needs in paediatric psychopharmacology: Present scenario and future perspectives. Eur. Neuropsychopharmacol. 2015, 25, 1513–1531. [Google Scholar] [CrossRef]
- Persico, A.M.; Ricciardello, A.; Lamberti, M.; Turriziani, L.; Cucinotta, F.; Brogna, C.; Vitiello, B.; Arango, C. The pediatric psychopharmacology of autism spectrum disorder: A systematic review—Part I: The past and the present. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 110, 110326. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Pharmacogenetics and psychiatric care: A review and commentary. J. Ment. Health Clin. Psychol. 2018, 2, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hudac, C.M.; Bove, J.; Barber, S.; Duyzend, M.; Wallace, A.; Martin, C.L.; Ledbetter, D.H.; Hanson, E.; Goin-Kochel, R.P.; Green-Snyder, L.A.; et al. Evaluating heterogeneity in ASD symptomatology, cognitive ability, and adaptive functioning among 16p11.2 CNV carriers. Autism Res. 2020, 13, 1300–1310. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.M.; Smith, R.; Lehman, C.; Mitchel, M.W.; Singer, K.; Weaver, W.C.; Chung, W. 16p11.2 Recurrent Deletion. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021; [updated 28 October 2021]. [Google Scholar]
- Jutla, A.; Turner, J.B.; Green Snyder, L.A.; Chung, W.K.; Veenstra-VanderWeele, J. Psychotic symptoms in 16p11.2 copy-number variant carriers. Autism Res. 2020, 13, 187–198. [Google Scholar] [CrossRef]
- Jacquemont, S.; Reymond, A.; Zufferey, F.; Harewood, L.; Walters, R.G.; Kutalik, Z.; Martinet, D.; Shen, Y.; Valsesia, A.; Beckmann, N.D.; et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011, 478, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Armando, M.; Ciampoli, M.; Padula, M.C.; Amminger, P.; De Crescenzo, F.; Maeder, J.; Schneider, M.; Schaer, M.; Managò, F.; Eliez, S.; et al. Favorable effects of omega-3 polyunsaturated fatty acids in attentional control and conversion rate to psychosis in 22q11.2 deletion syndrome. Neuropharmacology 2020, 168, 107995. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.; van der Gaag, M.; Nelson, B.; Smit, F.; Yuen, H.P.; Berger, M.; Krcmar, M.; French, P.; Amminger, G.P.; Bechdolf, A.; et al. Preventive interventions for individuals at ultra high risk for psychosis: An updated and extended meta-analysis. Clin. Psychol. Rev. 2021, 86, 102005. [Google Scholar] [CrossRef]
- Jobski, K.; Höfer, J.; Hoffmann, F.; Bachmann, C. Use of psychotropic drugs in patients with autism spectrum disorders: A systematic review. Acta Psychiatr. Scand. 2017, 135, 8–28. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Kim, E.; Cho, K.O.; Rothschild, A.; Sheng, M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron 1996, 17, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Irie, M.; Hata, Y.; Takeuchi, M.; Ichtchenko, K.; Toyoda, A.; Hirao, K.; Takai, Y.; Rosahl, T.W.; Südhof, T.C. Binding of neuroligins to PSD-95. Science 1997, 277, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Niethammer, M.; Rothschild, A.; Jan, Y.N.; Sheng, M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 1995, 378, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, H.J.; Fink, A.E.; Grant, S.G.; O’Dell, T.J. Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J. Physiol. 2008, 586, 5885–5900. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Hjälm, G.; Sakwe, A.M.; Engström, A.; Höglund, A.S.; Larsson, E.; Robinson, R.C.; Sundberg, C.; Rask, L. Selective interaction of megalin with postsynaptic density-95 (PSD-95)-like membrane-associated guanylate kinase (MAGUK) proteins. Biochem. J. 2003, 373 Pt 2, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Nithianantharajah, J.; Komiyama, N.H.; McKechanie, A.; Johnstone, M.; Blackwood, D.H.; St Clair, D.; Emes, R.D.; van de Lagemaat, L.N.; Saksida, L.M.; Bussey, T.J.; et al. Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nat. Neurosci. 2013, 16, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Yoo, T.; Kim, S.G.; Yang, S.H.; Kim, H.; Kim, E.; Kim, S.Y. A DLG2 deficiency in mice leads to reduced sociability and increased repetitive behavior accompanied by aberrant synaptic transmission in the dorsal striatum. Mol. Autism. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; McClellan, J.M.; McCarthy, S.E.; Addington, A.M.; Pierce, S.B.; Cooper, G.M.; Nord, A.S.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008, 320, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Egger, G.; Roetzer, K.M.; Noor, A.; Lionel, A.C.; Mahmood, H.; Schwarzbraun, T.; Boright, O.; Mikhailov, A.; Marshall, C.R.; Windpassinger, C.; et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 2014, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemany, S.; Ribasés, M.; Vilor-Tejedor, N.; Bustamante, M.; Sánchez-Mora, C.; Bosch, R.; Richarte, V.; Cormand, B.; Casas, M.; Ramos-Quiroga, J.A.; et al. New suggestive genetic loci and biological pathways for attention function in adult attention-deficit/hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 459–470. [Google Scholar] [CrossRef]
- Xing, J.; Kimura, H.; Wang, C.; Ishizuka, K.; Kushima, I.; Arioka, Y.; Yoshimi, A.; Nakamura, Y.; Shiino, T.; Oya-Ito, T.; et al. Resequencing and association analysis of six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci. Rep. 2016, 6, 27491. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, C.; Coppens, S.; Sekhara, T.; Dimov, I.; Pichon, B.; Lufin, N.; Addor, M.C.; Belligni, E.F.; Digilio, M.C.; Faletra, F.; et al. Novel promoters and coding first exons in DLG2 linked to developmental disorders and intellectual disability. Genome Med. 2017, 9, 67. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef] [Green Version]
- Noor, A.; Lionel, A.C.; Cohen-Woods, S.; Moghimi, N.; Rucker, J.; Fennell, A.; Thiruvahindrapuram, B.; Kaufman, L.; Degagne, B.; Wei, J.; et al. Copy number variant study of bipolar disorder in Canadian and UK populations implicates synaptic genes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 303–313. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387, 167–178. [Google Scholar] [CrossRef]
- Iacobucci, G.J.; Popescu, G.K. NMDA receptors: Linking physiological output to biophysical operation. Nat. Rev. Neurosci. 2017, 18, 236–249. [Google Scholar] [CrossRef]
- Farooq, M.; Kim, S.; Patel, S.; Khatri, L.; Hikima, T.; Rice, M.E.; Ziff, E.B. Lithium increases synaptic GluA2 in hippocampal neurons by elevating the δ-catenin protein. Neuropharmacology 2017, 113 Pt A, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Izsak, J.; Seth, H.; Iljin, M.; Theiss, S.; Ågren, H.; Funa, K.; Aigner, L.; Hanse, E.; Illes, S. Differential acute impact of therapeutically effective and overdose concentrations of lithium on human neuronal single cell and network function. Transl. Psychiatry 2021, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Roid, G.; Miller, L. Leiter international performance scale-revised: Examiner’s manual. In Leiter International Performance Scale-Revised; Roid, G.H., Miller, L.J., Eds.; Stoelting: Wood Dale, IL, USA, 1997. [Google Scholar]
- Biancardi, A.; Stoppa, E. The bells test revised: A proposal for the study of attention in childhood. Psichiatr. Dell’infanzia Dell’adolescenza 1997, 64, 73–84. [Google Scholar]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghali, N.; Sobey, G.; Burrows, N. Ehlers-Danlos syndromes. BMJ 2019, 366, l4966. [Google Scholar] [CrossRef] [PubMed]
- VanderJagt, K.; Butler, M.G. Ehlers-Danlos syndrome and other heritable connective tissue disorders that impact pregnancies can be detected using next-generation DNA sequencing. Arch. Gynecol. Obstet. 2019, 300, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. C. Semin. Med. Genet. 2015, 169C, 6–22. [Google Scholar] [CrossRef]
- Henneton, P.; Albuisson, J.; Adham, S.; Legrand, A.; Mazzella, J.M.; Jeunemaitre, X.; Frank, M. Accuracy of clinical diagnostic criteria for patients with vascular Ehlers-Danlos Syndrome in a tertiary referral centre. Circ. Genom. Precis. Med. 2019, 12, e001996. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H. Vascular Ehlers-Danlos Syndrome. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022; [updated 21 February 2019]. [Google Scholar]
- Rana, M.; Aziz, O.; Purkayastha, S.; Lloyd, J.; Wolfe, J.; Ziprin, P. Colonoscopic perforation leading to a diagnosis of Ehlers Danlos syndrome type IV: A case report and review of the literature. J. Med. Case. Rep. 2011, 5, 229. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.; Ricciardello, A.; Turriziani, L.; Calabrese, G.; Briguglio, M.; Boncoddo, M.; Bellomo, F.; Tomaiuolo, P.; Martines, S.; Bruschetta, M.; et al. FARP-1 deletion is associated with lack of response to autism treatment by Early Start Denver Model in a multiplex family. Mol. Genet. Genom. Med. 2020, 8, e1373. [Google Scholar] [CrossRef] [PubMed]
- Serret, S.; Thümmler, S.; Dor, E.; Vesperini, S.; Santos, A.; Askenazy, F. Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: Case reports. BMC Psychiatry 2015, 15, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egger, J.I.M.; Verhoeven, W.M.A.; Groenendijk-Reijenga, R.; Kant, S.G. Phelan-McDermid syndrome due to SHANK3 mutation in an intellectually disabled adult male: Successful treatment with lithium. BMJ Case Rep. 2017, 2017, bcr2017220778. [Google Scholar] [CrossRef]
- Butler, M.G.; Rafi, S.; Manzardo, A.M. High-resolution chromosome ideogram representation of currently recognized genes for autism spectrum disorders. Int. J. Molecul. Sci. 2015, 16, 6464–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| February 2017 | July 2017 | October 2017 | February 2018 | July 2018 | December 2019 | ||
|---|---|---|---|---|---|---|---|
| Leiter-R | Global I.Q. | 61 | 69 | 94 | 77 | 77 | 82 |
| Fluid Intelligence | 77 | 69 | 84 | 71 | 80 | 73 | |
| Brief I.Q. | 60 | 68 | 83 | 71 | 82 | 74 | |
| Bell test | Accuracy | −4.5 s.d. | −2.5 s.d. | −0.62 s.d. | - | - | - |
| Speed | −1.8 s.d. | −1.13 s.d. | +0.09 s.d. | - | - | - |
| EDS Subtype | Gene (Protein) | Inheritance |
|---|---|---|
| Classical EDS (cEDS) | Major: COL5A1 (COLLAGEN, TYPE V, α-1) Major: COL5A2 (COLLAGEN, TYPE V, α-2) Rare: COL1A1 (COLLAGEN, TYPE I, α-1) | AD |
| Classical-like EDS (clEDS) | TNXB (TENASCIN XB) | AR/AD? |
| Vascular EDS (vEDS) | Major: COL3A1 (COLLAGEN, TYPE III, α-1) Rare: COL1A1 (COLLAGEN, TYPE I, α-1) | AD |
| Kyphoscoliotic (kEDS) | PLOD1 (PROCOLLAGEN-LYSINE, 2-OXOGLUTARATE 5-DIOXYGENASE) FKBP14 (FK506-BINDING PROTEIN 14) | AR |
| Brittle cornea syndrome (BCS) | ZNF469 (ZINC FINGER PROTEIN 469) PRDM5 (PR DOMAIN-CONTAINING PROTEIN 5) | AR |
| Periodontal EDS (pEDS) | C1R (COMPLEMENT COMPONENT 1, r SUBCOMPONENT) C1S (COMPLEMENT COMPONENT 1, s SUBCOMPONENT) | AD |
| Anthrochalasia EDS (aEDS) | COL1A1 (COLLAGEN, TYPE I, α-1) COL1A2 (COLLAGEN, TYPE I, α-2) | AD |
| Musculocontractural EDS (mcEDS) | CHST14 (CARBOHYDRATE SULFOTRANSFERASE 14) DSE (DERMATAN SULFATE EPIMERASE) | AR |
| Myopathic EDS (mEDS) | COL12A1 (COLLAGEN, TYPE XII, α-1) | AD/AR |
| Cardiac-valvular EDS (cvEDS) | COL1A2 (COLLAGEN, TYPE I, α-2) | AR |
| Spondylodysplastic EDS (spEDS) | B4GALT7 (β-1,4-GALACTOSYLTRANSFERASE 7) B3GALT6 (β-1,3-GALACTOSYLTRANSFERASE 6) SLC39A13 (SOLUTE CARRIER FAMILY 39-ZINC TRANSPORTER, MEMBER 13) | AR |
| Dermatosparaxis EDS (dEDS) | ADAMTS2 (A DISINTEGRIN-LIKE AND METALLOPROTEINASE WITH THROMBOSPONDIN TYPE 1 MOTIF, 2) | AR |
| Hypermobile EDS (hEDS) | Unknown | AD? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, M.G.; Moreno-De-Luca, D.; Persico, A.M. Actionable Genomics in Clinical Practice: Paradigmatic Case Reports of Clinical and Therapeutic Strategies Based upon Genetic Testing. Genes 2022, 13, 323. https://doi.org/10.3390/genes13020323
Butler MG, Moreno-De-Luca D, Persico AM. Actionable Genomics in Clinical Practice: Paradigmatic Case Reports of Clinical and Therapeutic Strategies Based upon Genetic Testing. Genes. 2022; 13(2):323. https://doi.org/10.3390/genes13020323
Chicago/Turabian StyleButler, Merlin G., Daniel Moreno-De-Luca, and Antonio M. Persico. 2022. "Actionable Genomics in Clinical Practice: Paradigmatic Case Reports of Clinical and Therapeutic Strategies Based upon Genetic Testing" Genes 13, no. 2: 323. https://doi.org/10.3390/genes13020323