1. Introduction
Maize is an important staple food crop in sub-Saharan Africa (SSA) where a large area is under maize production [
1]. In east Africa, 82.48 million hectares (m ha) were covered by maize and about 156.21 million tons of maize grain were produced with productivity of 1.89 tons per ha (
http://www.fao.org/faostat/, accessed on 2 November 2021). Both biotic and abiotic stresses are the major threats to crop production, particularly maize in SSA. Drought stress, high costs of improved seeds and fertilizers [
2], and biotic stresses such as maize lethal necrosis (MLN) disease are the limiting factors for maize production in east Africa.
MLN was first reported in Kenya in 2011 and later reported in Tanzania, Uganda, Rwanda, D.R. Congo, and Ethiopia [
3,
4,
5].
Maize chlorotic mottle virus (MCMV) and
sugarcane mosaic virus (SCMV) viruses were the confirmed pathogens that have jointly incited the MLN disease [
5,
6,
7]. Both MCMV and SCMV are transmitted by insect vectors (MCMV by thrips and semipersistent beetles; SCMV by aphids) [
5,
8]. MCMV has been confirmed for its transmission by seeds and infected soils, making the management of MLN more challenging [
6,
9,
10,
11]. Based on the maize plant growth stages and environment conduciveness for MLN causing pathogens, the yield losses ranged from 30–100% [
12]. Thus, the management of MLN demands proper identification of resistant germplasm sources and associated genes or quantitative trait loci (QTL) that aid to develop the resistant hybrids or varieties [
13].
Doubled haploid (DH) lines allow complete homozygosity over lines developed through pedigree breeding; this allows precision in phenotyping over multiple locations and years [
14]. Further, high genetic variance in DH lines enhances response to selection [
15] by increasing heritability for various traits. Compared to breeding under well-watered (WW) conditions, the genetic variability, trait heritability, disease resistance, and selection gain are very low for breeding under water stress (WS) conditions [
16]; thus, WS condition makes the identification of best genotypes and expression of complex traits. These challenges are designed to be solved through established managed drought tolerance and disease screening facilities, not to lose the genetic variations, and to produce good yield under stress conditions. Understanding the maize crop’s behavior under WS for grain yield and yield-related traits, proper statistical design and breeding scheme help to select the best genotypes under WS environments [
16,
17].
Advancement in next-generation sequencing tools promoted genome-wide association studies (GWAS) in many crops including maize [
18]. Association analysis is based on the non-random association between genotypes and phenotypes of the diverse distantly related individuals [
19]. The significance of the marker–phenotype association could be declared when the marker polymorphism is located within the linkage disequilibrium (LD) region. To detect an association of complex traits, a minimum LD average with cut-off point of r
2 =0.1 was used [
19]. In maize, the rate of LD decay approximated to 1, 2, and 200–500 kb in landraces, diverse inbred lines, and commercial elite inbred lines, respectively [
20].
GWAS is useful in allele mining by dissecting the quantitative traits [
19]. QTL or gene mapping consists of linkage map construction and identifying genomic regions associated with the targeted QTL [
21]. QTL mapping helps to understand the genetic inheritance of quantitative traits [
22,
23]. Breeding for drought tolerance is complex since the trait is influenced by the environment and many genes with small effects [
24]. In maize, about 239 QTLs related to drought tolerance were reported [
25,
26]. Five drought-tolerant QTLs closely linked to grain yield were reported by Agrama and Moussa [
27]. Semagn et al. [
2] reported four meta-QTLs associated with grain yield for both under drought and optimum management. The high QTL detection power and fine resolution of mapping are exploited by joint linkage association mapping in multiple biparental populations [
28,
29,
30]. The identification and validation of novel genomic regions associated with economically important traits under WW and WS as well as MLN are important to accelerate the development of climate-resilient improved maize varieties to enhance high maize productions in smallholder families and contribute to food security [
12,
31,
32].
Genomic selection (GS) uses genome-wide markers to predict the breeding values of individuals by trapping the effects of both major and minor genes [
33]. In GS, from the training population, the effect of all markers are estimated, and then the genomic estimated breeding values (GEBVs) of the untested but genotyped lines are computed [
33]. Lines in the testing population are only genotyped, not phenotyped, and thus important in reducing the breeding cycle and increasing the genetic gain per unit time. GS is effective in several crops over a wide range of marker densities, trait complexities, and breeding populations [
34,
35,
36], where varying levels of prediction accuracy have been achieved in different studies.
To understand how WS affects grain yield and other key traits, this study was performed using a tropical maize population under drought and optimum conditions across multi-location field trials and the MLN effect under artificial inoculation in Kenya. The objectives of the study were to (i) evaluate the large set of 879 tropical and subtropical maize DH lines for their responses to MLN disease severity under artificial inoculation, grain yield (GY), and other yield-related traits under WW and WS conditions; (ii) identify genomic regions and putative candidate genes associated with these traits across the three management conditions; and (iii) assess the potential of GS within management conditions. This study will provide valuable information for uncovering the genetic basis of GY under WW and WS conditions.
4. Discussion
MLN is the major challenge to maize production in SSA, specifically in east African countries. CIMMYT in collaboration with national research institutions has developed resistance breeding strategies against MLN. A large number of maize genotypes were screened, and MLN disease-resistant source materials and resistance QTLs were identified to develop resistant varieties by integrating both conventional and molecular breeding techniques [
12,
31,
37,
38]. Nevertheless, searching additional MLN disease-resistant lines, evaluation of the genotype’s performance, identification and validation of QTLs associated with the target disease, GY, and other related traits play a vital role in the development of MLN disease-resistant varieties. In this study, 879 maize DH lines derived from 26 different populations were genotyped, and the performance of genotypes were evaluated under WW, WS, and MLN artificial inoculation management conditions. Among these 879 lines, 440 DH lines shared LapostaSeqC7 background lines as one of the parent, and the line LapostaSeqC7-F64 alone used as one of the parent to develop >250 DH lines, so, data was analyzed combinedly rather making it into subgroups based analyses.
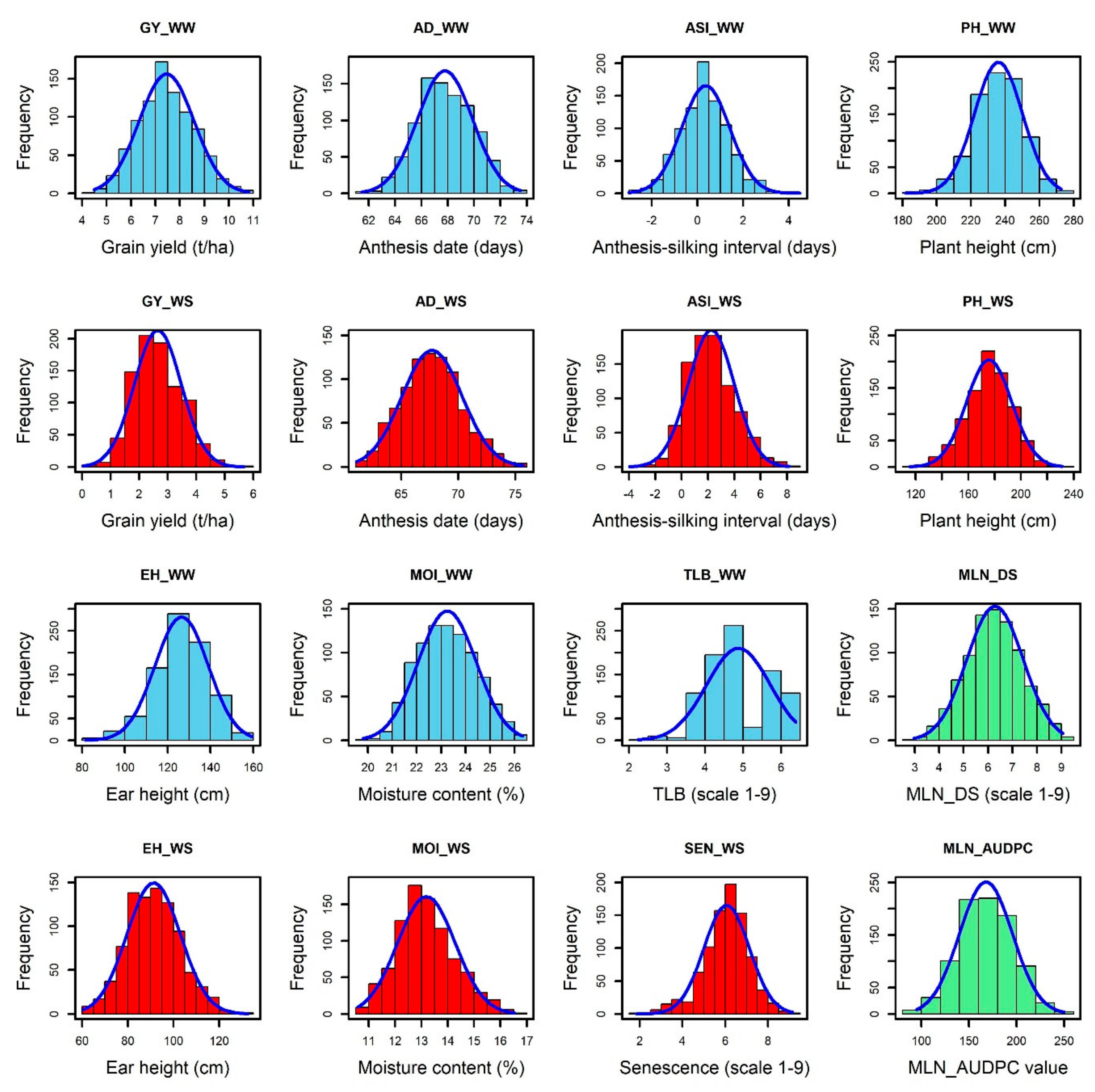
A significant genotype, genotype by environment interaction variances, and moderate to high broad-sense heritability were observed for GY and other related traits AD, ASI, PH, EH, TLB, MOI, and GLS measured under WW and WS conditions similar with the results reported by Yuan et al. [
24]. MLN-DS and AUDPC were highly heritable with 0.67 and 0.74, respectively, which is consistent with earlier reported studies [
31,
37,
53,
54]. Several genotypes have been evaluated by CIMMYT against MLN disease in search for resistant materials [
12,
24,
31,
32,
38,
55]; with the current study, we identified about 52 MLN disease resistant/tolerant genotypes while most of other genotypes were susceptible. Some of the maize genotypes with a score from 2 to 3 against MLN-DS were CKLMLN145667, CKLMLN145667, CKLMLN144135, CKLMLN145119, CKLMLN145173, CKLMLN143806, and CKLMLN143351, which could be selected as resistant materials to MLN disease. The mean performance of lines for GY was 7.54 t/ha and 2.7 t/ha under WW and WS environments, respectively, which has revealed a similar result in earlier study [
56]. The GY had positive correlations with both EH and PH and negative correlations with ASI and MOI under WW and WS management, respectively, which could help in an indirect selection for the GY under WW and WS conditions [
24].
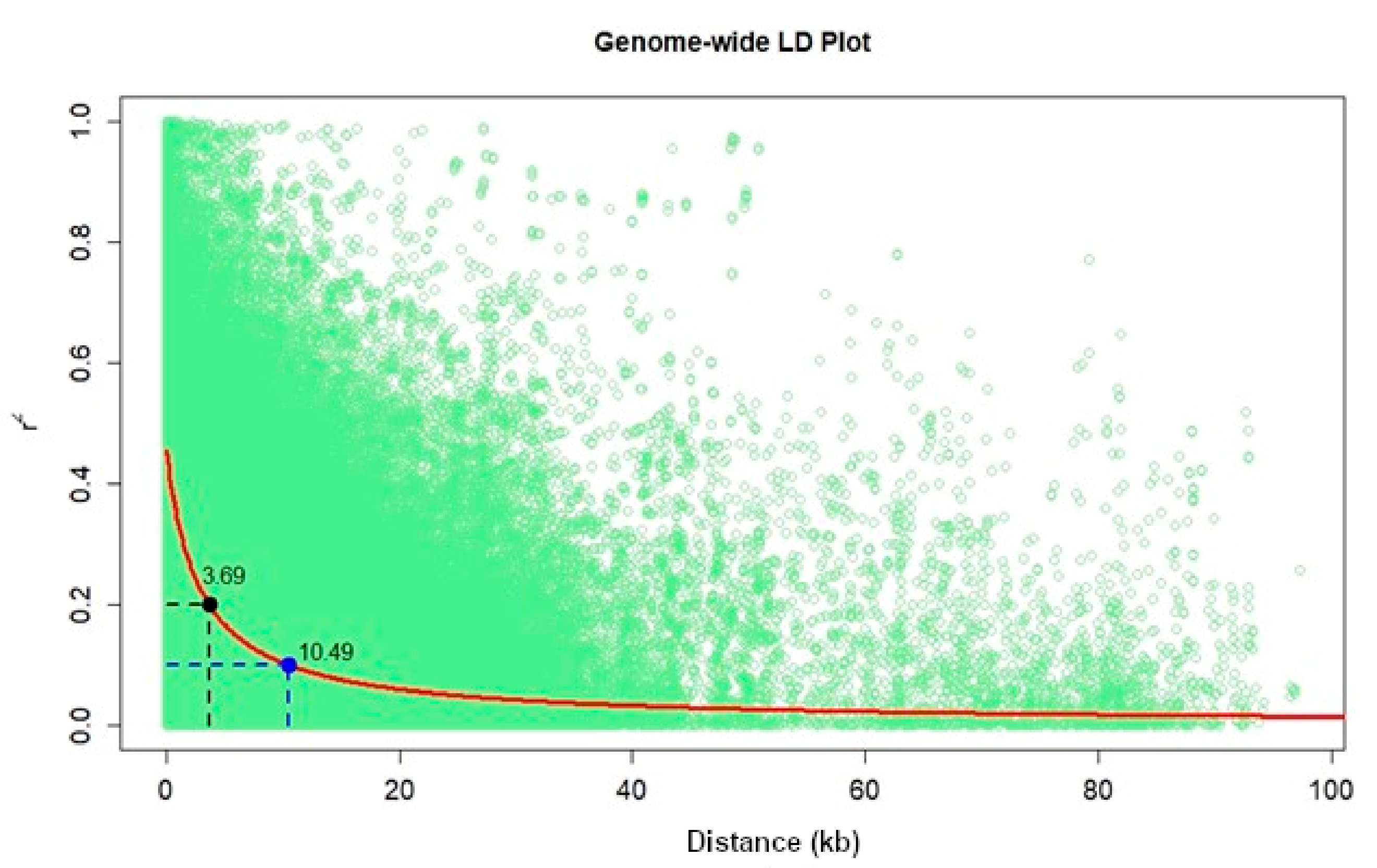
The number of SNPs required to achieve maximum mapping resolution depends on the magnitude of LD and LD decay with genetic distance [
57]. For GWAS, a large population is required since the LD or correlation between alleles in different genomic locations is generally based on the historical recombination between polymorphisms. In this study, we observed that the LD decay at r
2 = 0.1 and 0.2 cut-offs were 10.49 and 3.69 kb, respectively. Similarly, [
54] in the IMAS association panel also reported the genome-wide average LD decay of 14.97 kb at r
2 = 0.1 and 5.23 kb at r
2 = 0.2 [
54], and a similar range of LD decay was also reported by Rashid et al. [
58] in their association panel. LD decay in tropical maize germplasm was rapid compared to the temperate germplasm; possibly due to a broader genetic base, resulting from high recombination events [
59]. This provides an opportunity for breeders to select germplasm that integrates high GY with disease resistance and abiotic stress tolerance.
For population structure analyses, the Delta K line plot, principal component analyses, and population genetic distance relationship analyses suggested that the utilized DH populations are structured into three to four groups. In STRUCTURE, the optimum number of subgroups was determined based on the output log-likelihood of data (LnP (D. The peaks of the line plot (
Figure 4) suggest that the population could be divided into three or four distinct groups in order of possibility, with the K = 4 of delta K intersecting with LnP (D) showing a higher possibility. When K = 4, all lines were grouped as a mixed group and were further divided into three groups. The DH populations used in this study were grouped into CML395/CML505 derived DH lines, LaPostaSeq C7-F64 derived DH lines (174 individuals), and LaPostaSeq C7-F86 and LaPostaSeq F18 derived DH lines (265 individuals) (
Figure 4). Due to the inclusion of DH lines derive from crosses of selected inbred lines in the panel, we observed moderate structure in the present study. Several researchers also been reported moderate structure in the tropical maize germplasm [
29,
31,
37,
53,
54,
60].
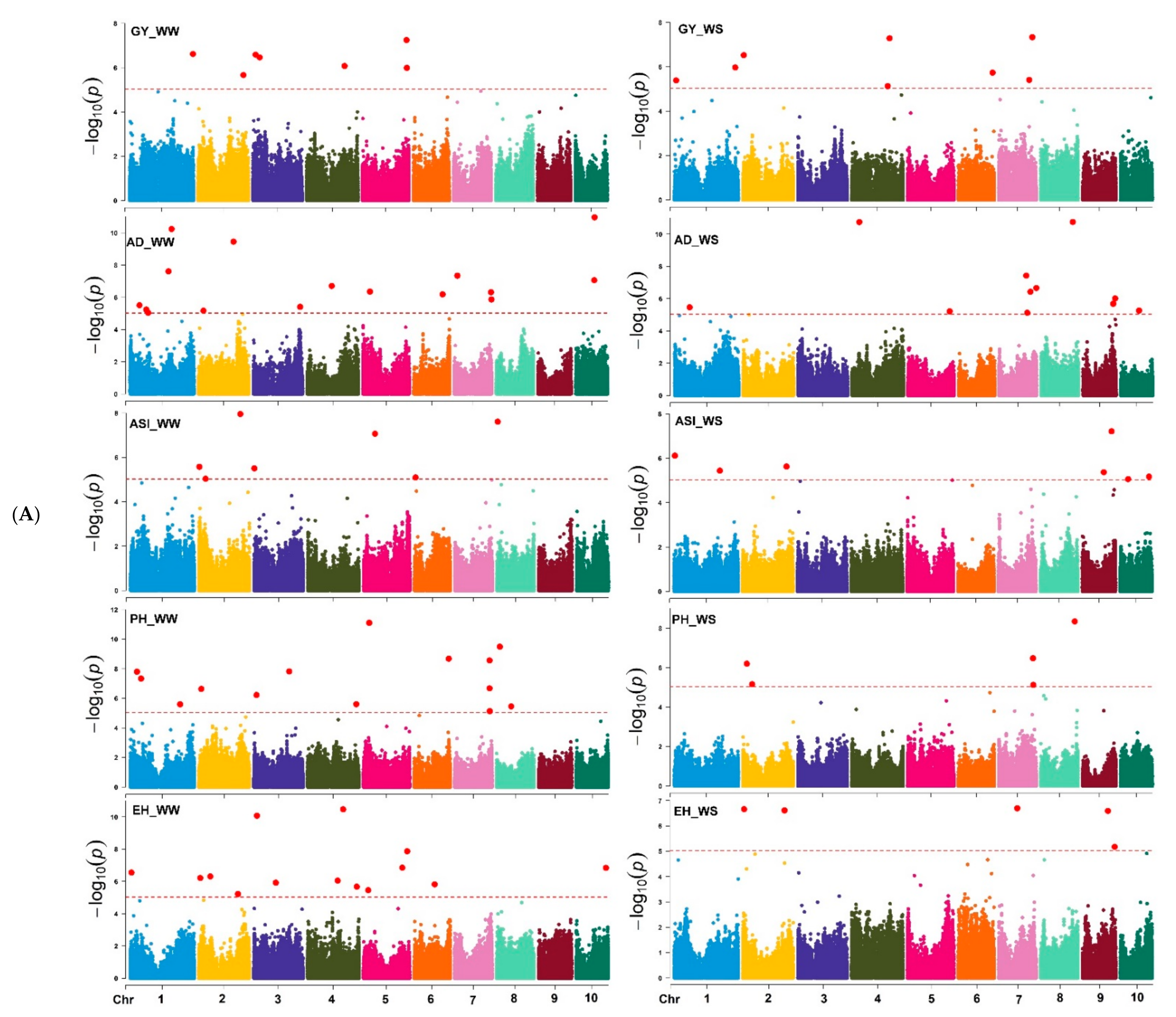
In this study, we identified the significant SNPs associated with target traits under WW, WS, and MLN artificial inoculations (
Table 3,
Table 4,
Table 5 and
Table 6). The results of this study for MLN-DS and AUDPC are similar to the reports in the biparental and DH population studied for the MLN-DS, AUDPC, and other traits genetic architecture [
12,
31,
38,
53,
54,
60]. Several putative candidate genes associated with the significant markers were identified for each of the studied traits (
Table 3,
Table 4,
Table 5 and
Table 6). For GY under WW, two putative candidate genes,
GRMZM2G017470 and
GRMZM2G030713, were identified, both located on chromosome 1 and, respectively, described as Dof zinc finger protein DOF3.6-like and O-fucosyltransferase 36 synthesis biological functions; whereas the candidate genes,
GRMZM2G472167 on chromosome 1 and
GRMZM2G019404 on chromosome 2, identified under WS were functionally described as peptide transporter PTR2 mha2 that involved in seed germination maternal control and plasma-membrane H+ATPase 2 that aid in activating secondary transport, respectively [
61,
62,
63]. These genes are more relevant to plants’ response to drought stress.
Putative candidate genes
GRMZM2G142383 and
GRMZM2G124136 detected for AD under WW and WS are functionally designated as Uridine kinase-like protein 2 chloroplastic involved in the pyrimidine salvage pathway [
64] and putative glycerol-3-phosphate transporter 4 involved in molecular function of transmembrane transporter activity [
65]. The SNPs
S8_3482389 and
S2_205904889 on chromosomes 8 and 2 were closely linked to ASI under both WW and WS associated with the putative candidate genes,
GRMZM2G136158 and
GRMZM2G105869, respectively. These candidate genes are involved in Peroxidase 24 that aid in responding to environmental stresses such as wounding, pathogen attack, and oxidative stress [
66], and histone-lysine
N-methyltransferase SUVR3 known to be involved in the development of pollen and female gametophyte, flowering, plant morphology, and the responses to stresses [
67], respectively.
The two important SNPs linked to PH
S6_161804186 under WW have shown a candidate gene
GRMZM2G170625, and
S2_43203188 under WS, which is located with the candidate gene,
GRMZM2G114523. Both designated candidate genes have been described as Jacalin-related lectin 3 and lysine histidine transporter-like 6 functions, respectively. Jacalin-related lectin 3 are proteins that bind carbohydrates and play an important role in plant development and resistance development to fungal pathogens [
68]. Lysine histidine transporter-like 6 helps to transport amino acid within or between the cells and is involved in plant uptake of amino acids [
69]. SNP,
S2_184012021 linked to EH under WW management was associated with the putative candidate gene,
GRMZM2G116196, that was described as AUGMIN subunit 5 (AUG5) essential for the development of gametophyte and sporophyte [
70] reproductions; another annotated gene
GRMZM2G365374 encoded as heat shock 70 kDa protein (HSPA1A) under WS was known to respond to heat-shock stress [
71].
The SNPs
S1_188031152 and
S8_11662494 associated with the MOI were detected with well-described putative candidate genes,
GRMZM2G419436 and
GRMZM2G700386, respectively. The gene
GRMZM2G419436 is characterized as well-associated receptor kinase 5 (WAK5), which significantly controls cell expansion, morphogenesis, and development [
72], while the
GRMZM2G700386 gene characterized as β-1,2-xylosyltransferase XYXT1 is involved in the xylosylation of xylan, the primary and secondary walls or major hemicellulose of angiosperms [
73]. The SNP,
S1_204865984, linked to SEN under WS environment was the annotated putative candidate gene,
GRMZM2G328309, explained as ribonuclease E/G-like protein, chloroplastic, which is a family of proteins that plays a pivotal function to metabolize RNA [
74].
The putative genes,
GRMZM2G009591 and
GRMZM2G101117, annotated from the SNPs
S1_246469847 and
S7_82649117 linked to GLS disease resistance had been characterized as pyrophosphate fructose 6-phosphate 1-phosphotransferase and GDSL esterase/lipase, respectively. The first gene,
GRMZM2G009591, is known to catalyze D-fructose 6-phosphate phosphorylation [
75]; the second gene,
GRMZM2G101117, is known for the molecular function hydrolytic activities of GDSL esterases and lipases enzymes [
76]. Under WW management, putative candidate genes,
GRMZM2G039173, GRMZM2G071023, and
GRMZM2G106119 were identified based on the associated SNPs
S6_157820129 and
S4_212595942 with the TLB resistance. Rédei [
77] has described the
GRMZM2G039173 gene as the major facilitator superfamily protein that aided in transporting small solutes based on the chemiosmotic ion gradients, while the second putative gene characterized by Chai et al. [
78] has functioned as a probable NAD kinase 2 chloroplast, which is actively involved in the protection of chloroplast against oxidative damage and synthesis of chlorophyll.
MLN-DS trait-associated SNPs
S3_184235364 and
S6_38115747 are annotated with
GRMZM2G429982 and
GRMZM5G818106 candidate genes that have osmotin-like protein and phospholipase A1-II 7 functions, respectively [
79,
80]. Kumar et al. [
80] characterized the candidate gene
GRMZM2G429982 as being involved in biotic and abiotic stresses tolerance in plants, whereas the candidate gene
GRMZM5G818106 has been described as protective of high temperature, cold, salt, and drought [
79]. Wu et al. [
81] reported that the function of the putative candidate gene
GRMZM2G003752, which was characterized as fasciclin-like arabinogalactan protein 10, was to respond to abiotic stress and mediate the growth and development of the plant. This candidate gene was annotated from the
S2_16652265 marker associated with AUDPC values. Similarly, the
S10_125845596 marker was linked to the AUDPC value and then the putative candidate gene,
GRMZM2G003917, was identified. This gene has been described by Wu et al. [
81] as a fasciclin-like arabinogalactan protein 7 (FLA7) gene responsible for the development of microspores and, under salt stress environment, maintaining proper plant cell expansion.
In the present study, a total of 98, 54, and 22 SNPs associated with various agronomic traits under WW, WS, and MLN conditions, respectively, were identified. Among these SNPs, some existed within different gene models whose genetic role is associated with either biotic or abiotic stress mechanisms. The favorable alleles can be identified by resequencing the detected candidate genes from contrasting, and these SNPs could be potentially converted to simple PCR-based markers to follow MAS in molecular breeding [
82]. Similarly, several GWAS studies reported large numbers of SNPs associated with important traits in maize [
83,
84].
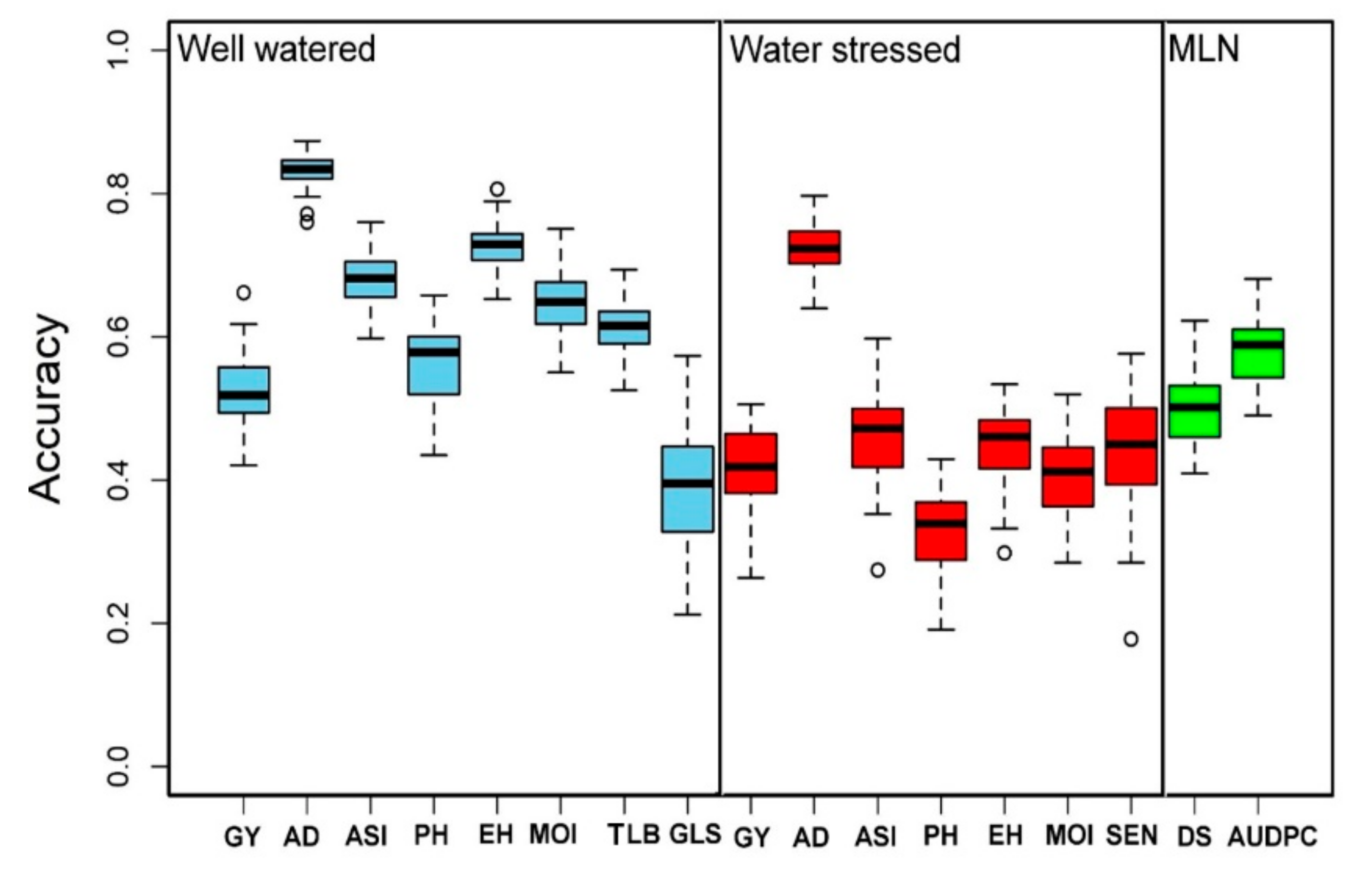
High genetic gain can be achieved for complex traits by integrating modern tools into maize breeding [
85,
86]. With several genotyping service providers available with a lower cost per sample and availability of advanced statistical models, genomic prediction is routinely applied in maize for several quantitative traits [
24,
85,
86]. In the present study, we compared the prediction accuracies under WW and WS conditions (
Figure 7). As expected for all the common traits measured in both WW and WS conditions, the prediction accuracies were slightly higher under WW conditions compared to WS conditions. The observed accuracy for all traits under WW, WS, and MLN conditions reveals the effect of heritability as the traits with higher heritability generally had higher prediction accuracy. The main factors affecting genomic prediction accuracy are the relationship between the training and testing populations, training population sizes, the population structure of training and testing sets, marker densities, genetic architecture and heritability of target traits, genotype by environment interactions, and statistical methods [
36,
62,
87,
88]. Knowing the genetic architecture of the target traits, it is possible to improve prediction accuracy while implementing GS [
35,
89]. Moderate-to-high accuracies observed in this study for the association panel offer promise in breeding for MLN and drought tolerance. The prediction accuracy of the association panel for MLN-DS and AUDPC is in agreement with earlier studies on MLN [
31] and MCMV [
38]. The prediction correlations observed for GY and other agronomic traits are equivalent to earlier studies reported in maize under different stresses [
24,
62,
85]. In GS, AD and ASI had higher accuracy compared to GY, which is expected, as these traits are less complex compared to GY [
24,
61,
62]. GWAS results revealed GY, and other agronomic traits evaluated under WW and WS conditions are complex in nature, controlled by many loci with minor effects, influenced by environmental factors. Therefore, they are difficult to track effectively in conventional breeding alone. Increase in prediction accuracy as well as increase in accumulation of favorable alleles with both minor and major effects is possible by integration of GS with GWAS results leads.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}