
Transcriptomics-Based Identification of Genes Related to Tapetum Degradation and Microspore Development in Lily


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Lily Anther Histological Observation
2.3. RNA Extraction and Library Construction
2.4. Error Correction of PacBio Iso-Seq Full-Length cDNA Reads
2.5. Gene Prediction and Annotation
2.6. Quantification of Transcripts Using Illumina Short Reads
2.7. The ‘eFP-Seq’ Browser Analysis of DEGs
2.8. Quantitative Real-Time PCR
3. Results
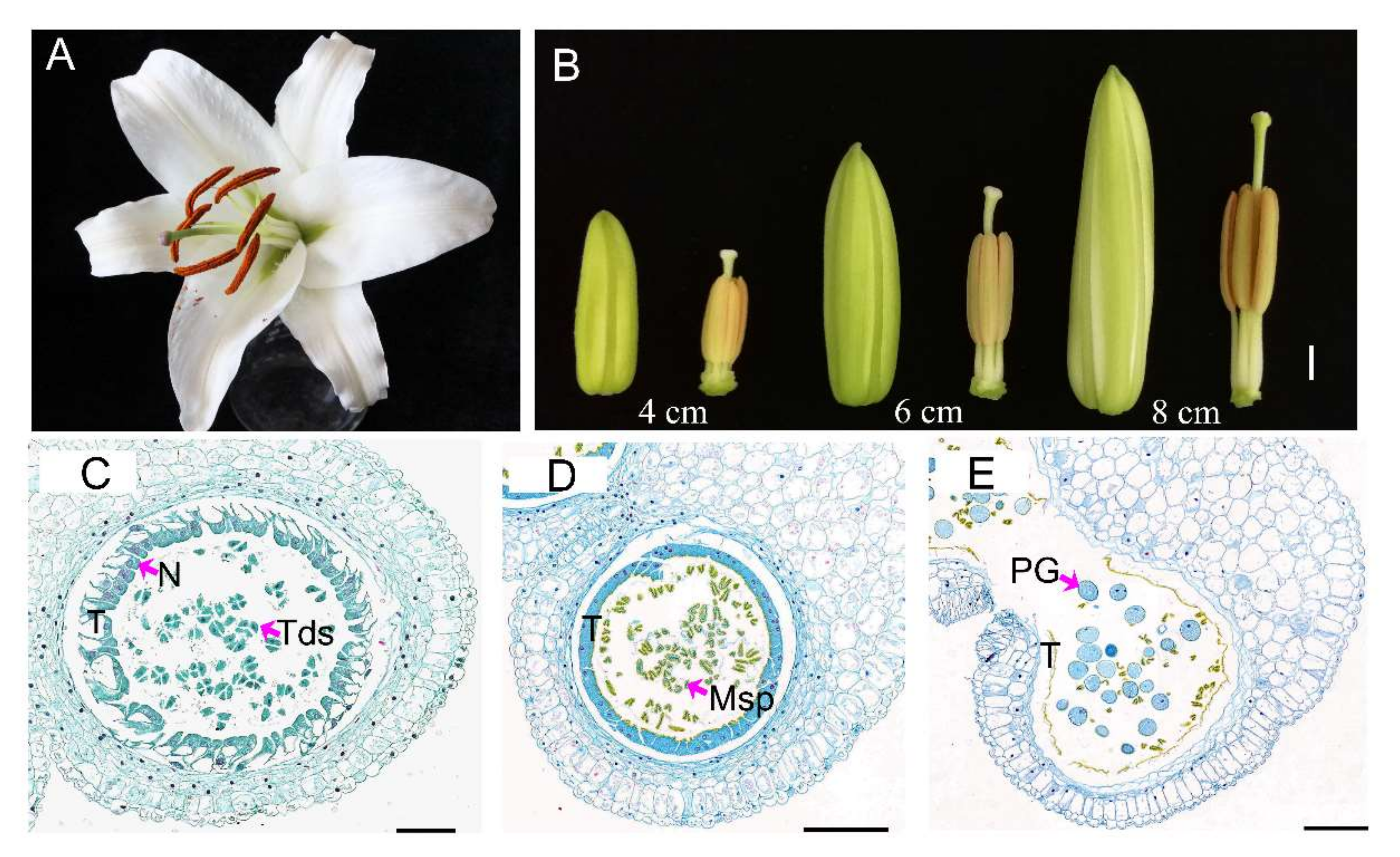
3.1. Lily Anthers Cytology Observation
3.2. Overall Analysis of Transcriptome Sequencing and Gene Annotation
3.3. Screening of Differentially Expressed Genes (DEGs) and Co-Expression Analysis
3.4. GO Enrichment and KEGG Pathway Analysis
3.5. Analysis of Transcription Factors Related to Tapetum Degradation and Microspore Development
3.6. Identification of DEGs Related to Tapetum Degradation and Microspore Development
3.7. Differential Gene Expression Validation by qRT-PCR Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sui, J.; Cao, X.; Yi, M.; Wu, J.; He, J. Isolation and characterization of LoAMS gene in anther development of lily (Lilium oriental hybrids). N. Z. J. Crop. Hortic. Sci. 2020, 48, 257–269. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Z.; Wang, L.; Wu, M.; Zhang, D.; Fang, W.; Chen, F.; Teng, N. Cytological and molecular characteristics of pollen abortion in lily with dysplastic tapetum. Hortic. Plant J. 2019, 5, 281–294. [Google Scholar] [CrossRef]
- Feng, J.; Wu, Z.; Wang, X.; Zhang, Y.; Teng, N. Analysis of Pollen Allergens in Lily by Transcriptome and Proteome Data. Int. J. Mol. Sci. 2019, 20, 5892. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; He, J.; Wu, J.; Gong, B.; Cao, X.; Seng, S.; Wu, Z.; Wu, C.; Liu, C.; Yi, M. Characterization and functional analysis of transcription factor lomyb80 related to anther development in lily (Lilium oriental hybrids). J. Plant Growth Regul. 2015, 34, 545–557. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Beals, T.P.; Sanders, P.M. Anther development: Basic principles and practical applications. Plant Cell 1993, 5, 1217. [Google Scholar]
- Zhang, S.; Fang, Z.; Zhu, J.; Gao, J.; Yang, Z. OsMYB103 is required for rice anther development by regulating tapetum development and exine formation. Chin. Sci. Bull. 2010, 55, 3288–3297. [Google Scholar] [CrossRef]
- Mariani, C.; De Beuckeleer, M.; Truettner, J.; Leemans, J.; Goldberg, R.B. Induction of male sterility in plants by a chimaeric ribonuclease gene. Nature 1990, 347, 737–741. [Google Scholar] [CrossRef]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Timofejeva, L.; Chen, C.; Grossniklaus, U.; Ma, H. Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development 2006, 133, 3085–3095. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, H.; Li, H.; Gao, J.F.; Jiang, H.; Wang, C.; Guan, Y.F.; Yang, Z.N. Defective in Tapetal development and function 1 is essential for anther development and tapetal function for microspore maturation in Arabidopsis. Plant J. 2008, 55, 266–277. [Google Scholar] [CrossRef]
- Sorensen, A.M.; Kröber, S.; Unte, U.S.; Huijser, P.; Dekker, K.; Saedler, H. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 2003, 33, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, C.; Yuan, Z.; Zhang, D.; Gondwe, M.Y.; Ding, Z.; Liang, W.; Zhang, D.; Wilson, Z.A. The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell 2010, 22, 91–107. [Google Scholar] [CrossRef]
- Ferguson, A.C.; Pearce, S.; Band, L.R.; Yang, C.; Ferjentsikova, I.; King, J.; Yuan, Z.; Zhang, D.; Wilson, Z.A. Biphasic regulation of the transcription factor ABORTED MICROSPORES (AMS) is essential for tapetum and pollen development in Arabidopsis. New Phytol. 2017, 213, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.B.; Zhu, J.; Gao, J.F.; Wang, C.; Li, H.; Li, H.; Zhang, H.Q.; Zhang, S.; Wang, D.M.; Wang, Q.X. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis. Plant J. 2007, 52, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, G.; Chang, Y.; Li, X.; Yang, J.; Huang, X.; Yu, Q.; Chen, H.; Wu, T.; Yang, Z. AtMYB103 is a crucial regulator of several pathways affecting Arabidopsis anther development. Sci. China Life Sci. 2010, 53, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Wilson, Z.A.; Morroll, S.M.; Dawson, J.; Swarup, R.; Tighe, P.J. The Arabidopsis MALE STERILITY1 (MS1) gene is a transcriptional regulator of male gametogenesis, with homology to the PHD-finger family of transcription factors. Plant J. 2001, 28, 27–39. [Google Scholar] [CrossRef]
- Yang, C.; Vizcay-Barrena, G.; Conner, K.; Wilson, Z.A. MALE STERILITY1 is required for tapetal development and pollen wall biosynthesis. Plant Cell 2007, 19, 3530–3548. [Google Scholar] [CrossRef]
- Huang, G.; Yi, Q.; Tong, L.; Hui, Y.; Aide, W.; Dongmei, T. Comparative transcriptome analysis of actinidia arguta fruits reveals the involvement of various transcription factors in ripening. Hortic. Plant J. 2018, 4, 35–42. [Google Scholar] [CrossRef]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sullivan, A.; Purohit, P.K.; Freese, N.H.; Pasha, A.; Esteban, E.; Waese, J.; Wu, A.; Chen, M.; Chin, C.Y.; Song, R. An ‘eFP-Seq Browser’for visualizing and exploring RNA sequencing data. Plant J. 2019, 100, 641–654. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lutgen, D.; Ritter, R.; Olsen, R.A.; Schielzeth, H.; Gruselius, J.; Ewels, P.; García, J.T.; Shirihai, H.; Schweizer, M.; Suh, A. Linked-read sequencing enables haplotype-resolved resequencing at population scale. Mol. Ecol. Resour. 2020, 20, 1311–1322. [Google Scholar] [CrossRef]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Weigt, D.; Niemann, J.; Siatkowski, I.; Zyprych-Walczak, J.; Olejnik, P.; Kurasiak-Popowska, D. Effect of zearalenone and hormone regulators on microspore embryogenesis in anther culture of wheat. Plants 2019, 8, 487. [Google Scholar] [CrossRef] [PubMed]
- Verma, N. Transcriptional regulation of anther development in Arabidopsis. Gene 2019, 689, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Drews, G.N.; Meyerowitz, E.M. Expression of the Arabidopsis floral homeotic gene AGAMOUS is restricted to specific cell types late in flower development. Plant Cell 1991, 3, 749–758. [Google Scholar] [PubMed]
- Sieburth, L.E.; Meyerowitz, E.M. Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell 1997, 9, 355–365. [Google Scholar]
- Ito, T.; Ng, K.-H.; Lim, T.-S.; Yu, H.; Meyerowitz, E.M. The homeotic protein AGAMOUS controls late stamen development by regulating a jasmonate biosynthetic gene in Arabidopsis. Plant Cell 2007, 19, 3516–3529. [Google Scholar] [CrossRef]
- Higginson, T.; Li, S.F.; Parish, R.W. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. 2003, 35, 177–192. [Google Scholar] [CrossRef]
- Alonso-Peral, M.M.; Li, J.; Li, Y.; Allen, R.S.; Schnippenkoetter, W.; Ohms, S.; White, R.G.; Millar, A.A. The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 2010, 154, 757–771. [Google Scholar] [CrossRef]
- Liu, L.; Fan, X.-d. Tapetum: Regulation and role in sporopollenin biosynthesis in Arabidopsis. Plant Mol. Biol. 2013, 83, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, J.; Sun, L.; Hu, Y.; Yu, J.; Wang, C.; Zhang, F.; Hou, H.; Liang, W.; Zhang, D. Two rice MYB transcription factors maintain male fertility in response to photoperiod by modulating sugar partitioning. New Phytol. 2021, 231, 1612–1629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liang, W.; Yang, X.; Luo, X.; Jiang, N.; Ma, H.; Zhang, D. Carbon starved anther encodes a MYB domain protein that regulates sugar partitioning required for rice pollen development. Plant Cell 2010, 22, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Parish, R.W.; Li, S.F. Death of a tapetum: A programme of developmental altruism. Plant Sci. 2010, 178, 73–89. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Z.; Vizcay-Barrena, G.; Shi, J.; Liang, W.; Yuan, Z.; Werck-Reichhart, D.; Schreiber, L.; Wilson, Z.A.; Zhang, D. ABORTED MICROSPORES acts as a master regulator of pollen wall formation in Arabidopsis. Plant Cell 2014, 26, 1544–1556. [Google Scholar] [CrossRef]
- Guo, J.; Liu, C.; Wang, P.; Cheng, Q.; Sun, L.; Yang, W.; Shen, H. The aborted microspores (AMS)-like gene is required for anther and microspore development in pepper (Capsicum annuum L.). Int. J. Mol. Sci. 2018, 19, 1341. [Google Scholar] [CrossRef]
- Fiorio, M.; Gambarin, M.; Valente, E.M.; Liberini, P.; Loi, M.; Cossu, G.; Moretto, G.; Bhatia, K.P.; Defazio, G.; Aglioti, S.M. Defective temporal processing of sensory stimuli in DYT1 mutation carriers: A new endophenotype of dystonia? Brain 2007, 130, 134–142. [Google Scholar] [CrossRef]
- Li, N.; Zhang, D.S.; Liu, H.S.; Yin, C.S.; Li, X.X.; Liang, W.Q.; Yuan, Z.; Xu, B.; Chu, H.W.; Wang, J.; et al. The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef]
- Niu, N.; Liang, W.; Yang, X.; Jin, W.; Wilson, Z.A.; Hu, J.; Zhang, D. EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice. Nat. Commun. 2013, 4, 1445. [Google Scholar] [CrossRef]
- Li, H.; Yuan, Z.; Vizcay-Barrena, G.; Yang, C.; Liang, W.; Zong, J.; Wilson, Z.A.; Zhang, D. PERSISTENT TAPETAL CELL1 encodes a PHD-finger protein that is required for tapetal cell death and pollen development in rice. Plant Physiol. 2011, 156, 615–630. [Google Scholar] [CrossRef]
- Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. The NAC transcription factors NST1 and NST2 of Arabidopsis regulate secondary wall thickenings and are required for anther dehiscence. Plant Cell 2005, 17, 2993–3006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, F.; Zhong, Y.; He, J.; Li, L. Modulation of NAC transcription factor NST1 activity by XYLEM NAC DOMAIN1 regulates secondary cell wall formation in Arabidopsis. J. Exp. Bot. 2020, 71, 1449–1458. [Google Scholar] [CrossRef]
- Li, X.; Gao, S.; Tang, Y.; Li, L.; Zhang, F.; Feng, B.; Fang, Z.; Ma, L.; Zhao, C. Genome-wide identification and evolutionary analyses of bZIP transcription factors in wheat and its relatives and expression profiles of anther development related TabZIP genes. BMC Genom. 2015, 16, 976. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Guan, Y.; Ren, H.; Zhang, F.; Chen, F. A bZIP transcription factor, OsABI5, is involved in rice fertility and stress tolerance. Plant Mol. Biol. 2008, 66, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Vizcay-Barrena, G.; Wilson, Z.A. Altered tapetal PCD and pollen wall development in the Arabidopsis ms1 mutant. J. Exp. Bot. 2006, 57, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, Z.; Cao, J. Pollen wall development: The associated enzymes and metabolic pathways. Plant Biol. 2013, 15, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-D.; Xue, J.-S.; Zhu, J.; Yang, Z.-N. Gene regulatory network for tapetum development in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 1559. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xun, Q.; Guo, Y.; Zhang, J.; Cheng, K.; Shi, T.; He, K.; Hou, S.; Gou, X.; Li, J. Genome-wide expression pattern analyses of the Arabidopsis leucine-rich repeat receptor-like kinases. Mol. Plant 2016, 9, 289–300. [Google Scholar] [CrossRef]
- Ge, X.; Chang, F.; Ma, H. Signaling and transcriptional control of reproductive development in Arabidopsis. Curr. Biol. 2010, 20, R988–R997. [Google Scholar] [CrossRef]
- Yang, L.; Qian, X.; Chen, M.; Fei, Q.; Meyers, B.C.; Liang, W.; Zhang, D. Regulatory role of a receptor-like kinase in specifying anther cell identity. Plant Physiol. 2016, 171, 2085–2100. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, D. The role of receptor-like kinases in regulating plant male reproduction. Plant Reprod. 2018, 31, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Szakmary, A.; Cox, D.N.; Wang, Z.; Lin, H. Regulatory relationship among piwi, pumilio, and bag-of-marbles in Drosophila germline stem cell self-renewal and differentiation. Curr. Biol. 2005, 15, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Hart, E.M.; Gupta, M.; Wühr, M.; Silhavy, T.J. The synthetic phenotype of Δ bamB Δ bamE double mutants results from a lethal jamming of the bam complex by the lipoprotein RcsF. MBio 2019, 10, e00662-19. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Aya, K.; Hobo, T.; Sakakibara, H.; Kojima, M.; Shim, R.A.; Hasegawa, Y.; Ueguchi-Tanaka, M.; Matsuoka, M. Comprehensive transcriptome analysis of phytohormone biosynthesis and signaling genes in microspore/pollen and tapetum of rice. Plant Cell Physiol. 2008, 49, 1429–1450. [Google Scholar] [CrossRef]
- Acosta, I.F.; Przybyl, M. Jasmonate signaling during Arabidopsis stamen maturation. Plant Cell Physiol. 2019, 60, 2648–2659. [Google Scholar] [CrossRef]
- Shivanna, K.R. Pollen Biology and Biotechnology; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Sanders, P.M.; Bui, A.Q.; Weterings, K.; McIntire, K.; Hsu, Y.-C.; Lee, P.Y.; Truong, M.T.; Beals, T.; Goldberg, R. Anther developmental defects in Arabidopsis thaliana male-sterile mutants. Sex. Plant Reprod. 1999, 11, 297–322. [Google Scholar] [CrossRef]
- Stieglitz, H.; Stern, H. Regulation of β-1, 3-glucanase activity in developing anthers of Lilium. Dev. Biol. 1973, 34, 169–173. [Google Scholar] [CrossRef]
- Wan, L.; Zha, W.; Cheng, X.; Liu, C.; Lv, L.; Liu, C.; Wang, Z.; Du, B.; Chen, R.; Zhu, L. A rice β-1, 3-glucanase gene Osg1 is required for callose degradation in pollen development. Planta 2011, 233, 309–323. [Google Scholar] [CrossRef]
- Pacini, E. Relationships between tapetum, loculus, and pollen during development. Int. J. Plant Sci. 2010, 171, 1–11. [Google Scholar] [CrossRef]
- Walbot, V.; Egger, R.L. Pre-meiotic anther development: Cell fate specification and differentiation. Annu. Rev. Plant Biol. 2016, 67, 365–395. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, L. Specification of tapetum and microsporocyte cells within the anther. Curr. Opin. Plant Biol. 2014, 17, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wijeratne, A.J.; Tang, C.; Zhang, T.; Fenelon, R.E.; Owen, H.A.; Zhao, D. Ectopic expression of TAPETUM DETERMINANT1 affects ovule development in Arabidopsis. J. Exp. Bot. 2016, 67, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-L.; Jiang, L.; Puah, C.S.; Xie, L.-F.; Zhang, X.-Q.; Chen, L.-Q.; Yang, W.-C.; Ye, D. Overexpression of TAPETUM DETERMINANT1 alters the cell fates in the Arabidopsis carpel and tapetum via genetic interaction with excess microsporocytes1/extra sporogenous cells. Plant Physiol. 2005, 139, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Tang, D.; Shen, Y.; Hu, Q.; Wang, K.; Li, M.; Lu, T.; Cheng, Z. MIL2 (MICROSPORELESS2) regulates early cell differentiation in the rice anther. New Phytol. 2012, 196, 402–413. [Google Scholar] [CrossRef]
- Zhao, X.; de Palma, J.; Oane, R.; Gamuyao, R.; Luo, M.; Chaudhury, A.; Hervé, P.; Xue, Q.; Bennett, J. OsTDL1A binds to the LRR domain of rice receptor kinase MSP1, and is required to limit sporocyte numbers. Plant J. 2008, 54, 375–387. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, L.; Lu, Y.; Wu, Y.; Dumenil, J.; Zhu, Z.; Bevan, M.W.; Li, Y. The ubiquitin receptors DA1, DAR1, and DAR2 redundantly regulate endoreduplication by modulating the stability of TCP14/15 in Arabidopsis. Plant Cell 2015, 27, 649–662. [Google Scholar] [CrossRef]
- Safavian, D.; Jamshed, M.; Sankaranarayanan, S.; Indriolo, E.; Samuel, M.A.; Goring, D.R. High humidity partially rescues the Arabidopsis thaliana exo70A1 stigmatic defect for accepting compatible pollen. Plant Reprod. 2014, 27, 121–127. [Google Scholar] [CrossRef]
- Wan, L.; Xia, X.; Hong, D.; Li, J.; Yang, G. Abnormal vacuolization of the tapetum during the tetrad stage is associated with male sterility in the recessive genic male sterile Brassica napus L. Line 9012A. J. Plant Biol. 2010, 53, 121–133. [Google Scholar] [CrossRef]
- Singh, P.; Rao, R.N.; Reddy, J.R.C.; Prasad, R.; Kotturu, S.K.; Ghosh, S.; Mukhopadhyay, S. PE11, a PE/PPE family protein of Mycobacterium tuberculosis is involved in cell wall remodeling and virulence. Sci. Rep. 2016, 6, 1–16. [Google Scholar]
- Jaquinod, M.; Villiers, F.; Kieffer-Jaquinod, S.; Hugouvieux, V.; Bruley, C.; Garin, J.; Bourguignon, J. A proteomics dissection of Arabidopsis thaliana vacuoles isolated from cell culture. Mol. Cell. Proteom. 2007, 6, 394–412. [Google Scholar] [CrossRef]
- Rodrıguez-Navarro, S.; Fischer, T.; Luo, M.-J.; Antúnez, O.; Brettschneider, S.; Lechner, J.; Pérez-Ortín, J.E.; Reed, R.; Hurt, E. Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell 2004, 116, 75–86. [Google Scholar] [CrossRef]
- Sarthy, A.V.; McGonigal, T.; Coen, M.; Frost, D.J.; Meulbroek, J.A.; Goldman, R.C. Phenotype in Candida albicans of a disruption of the BGL2 gene encoding a 1, 3-β-glucosyltransferase. Microbiology 1997, 143, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Furumizu, C.; Sawa, S. Insight into early diversification of leucine-rich repeat receptor-like kinases provided by the sequenced moss and hornwort genomes. Plant Mol. Biol. 2021, 107, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Laohavisit, A.; Wakatake, T.; Ishihama, N.; Mulvey, H.; Takizawa, K.; Suzuki, T.; Shirasu, K. Quinone perception in plants via leucine-rich-repeat receptor-like kinases. Nature 2020, 587, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.; Schmidt, E.D.; Bakker, F.T. Leucine-rich repeat receptor-like kinase II phylogenetics reveals five main clades throughout the plant kingdom. Plant J. 2020, 103, 547–560. [Google Scholar] [CrossRef]
- Crook, A.D.; Willoughby, A.C.; Hazak, O.; Okuda, S.; Van Der Molen, K.R.; Soyars, C.L.; Cattaneo, P.; Clark, N.M.; Sozzani, R.; Hothorn, M. BAM1/2 receptor kinase signaling drives CLE peptide-mediated formative cell divisions in Arabidopsis roots. Proc. Natl. Acad. Sci. USA 2020, 117, 32750–32756. [Google Scholar] [CrossRef]
- Chen, W.; Lv, M.; Wang, Y.; Wang, P.-A.; Cui, Y.; Li, M.; Wang, R.; Gou, X.; Li, J. BES1 is activated by EMS1-TPD1-SERK1/2-mediated signaling to control tapetum development in Arabidopsis thaliana. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Zhao, D. Control of anther cell differentiation: A teamwork of receptor-like kinases. Sex. Plant Reprod. 2009, 22, 221–228. [Google Scholar] [CrossRef]
- Jia, G.; Liu, X.; Owen, H.A.; Zhao, D. Signaling of cell fate determination by the TPD1 small protein and EMS1 receptor kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 2220–2225. [Google Scholar] [CrossRef]
- Mizuno, S.; Osakabe, Y.; Maruyama, K.; Ito, T.; Osakabe, K.; Sato, T.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Receptor-like protein kinase 2 (RPK 2) is a novel factor controlling anther development in Arabidopsis thaliana. Plant J. 2007, 50, 751–766. [Google Scholar] [CrossRef]
- Nonomura, K.; Miyoshi, K.; Eiguchi, M.; Suzuki, T.; Miyao, A.; Hirochika, H.; Kurata, N. The MSP1 gene is necessary to restrict the number of cells entering into male and female sporogenesis and to initiate anther wall formation in rice. Plant Cell 2003, 15, 1728–1739. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Euring, D.; Cha, J.Y.; Lin, Z.; Lu, M.; Huang, L.-J.; Kim, W.Y. Plant hormone-mediated regulation of heat tolerance in response to global climate change. Front. Plant Sci. 2021, 11, 2318. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zou, Y.; Kaw, H.Y.; Wang, G.; Sun, H.; Cai, L.; Li, C.; Meng, L.-Y.; Li, D. Recent developments and emerging trends of mass spectrometric methods in plant hormone analysis: A review. Plant Methods 2020, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, M.A.; Nelson, D.C.; Weijers, D. Evolution of plant hormone response pathways. Annu. Rev. Plant Biol. 2020, 71, 327–353. [Google Scholar] [CrossRef]
- Kovaleva, L.; Voronkov, A.; Zakharova, E.; Andreev, I. ABA and IAA control microsporogenesis in Petunia hybrida L. Protoplasma 2018, 255, 751–759. [Google Scholar] [CrossRef]
- Jewell, J.B.; Browse, J. Epidermal jasmonate perception is sufficient for all aspects of jasmonate-mediated male fertility in Arabidopsis. Plant J. 2016, 85, 634–647. [Google Scholar] [CrossRef]
- Bassa, C.; Mila, I.; Bouzayen, M.; Audran-Delalande, C. Phenotypes associated with down-regulation of Sl-IAA27 support functional diversity among Aux/IAA family members in tomato. Plant Cell Physiol. 2012, 53, 1583–1595. [Google Scholar] [CrossRef]
- Keller, M.; Schleiff, E.; Simm, S. miRNAs involved in transcriptome remodeling during pollen development and heat stress response in Solanum lycopersicum. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Richter, S.; Müller, L.M.; Stierhof, Y.-D.; Mayer, U.; Takada, N.; Kost, B.; Vieten, A.; Geldner, N.; Koncz, C.; Jürgens, G. Polarized cell growth in Arabidopsis requires endosomal recycling mediated by GBF1-related ARF exchange factors. Nat. Cell Biol. 2012, 14, 80–86. [Google Scholar] [CrossRef]
- Yao, X.; Chen, J.; Zhou, J.; Yu, H.; Ge, C.; Zhang, M.; Gao, X.; Dai, X.; Yang, Z.-N.; Zhao, Y. An essential role for miRNA167 in maternal control of embryonic and seed development. Plant Physiol. 2019, 180, 453–464. [Google Scholar] [CrossRef]
- Gavassi, M.A.; Silva, G.S.; da Silva, C.d.M.S.; Thompson, A.J.; Macleod, K.; Oliveira, P.M.R.; Cavalheiro, M.F.; Domingues, D.S.; Habermann, G. NCED expression is related to increased ABA biosynthesis and stomatal closure under aluminum stress. Environ. Exp. Bot. 2021, 185, 104404. [Google Scholar] [CrossRef]
- Qi, L.; Liu, S.; Li, C.; Fu, J.; Jing, Y.; Cheng, J.; Li, H.; Zhang, D.; Wang, X.; Dong, X. PHYTOCHROME-INTERACTING FACTORS interact with the ABA receptors PYL8 and PYL9 to orchestrate ABA signaling in darkness. Mol. Plant 2020, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, B.; Geng, S.; Arellano, C.; Chen, S.; Qu, R. Effects of overexpression of jasmonic acid biosynthesis genes on nicotine accumulation in tobacco. Plant Direct 2018, 2, e00036. [Google Scholar] [CrossRef] [PubMed]
- Vidhyasekaran, P. Gibberellin Signaling in Plant Innate Immunity. In Plant Hormone Signaling Systems in Plant Innate Immunity; Springer: Berlin/Heidelberg, Germany, 2015; pp. 383–401. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sui, J.; Jia, W.; Xin, Y.; Zhang, Y. Transcriptomics-Based Identification of Genes Related to Tapetum Degradation and Microspore Development in Lily. Genes 2022, 13, 366. https://doi.org/10.3390/genes13020366
Sui J, Jia W, Xin Y, Zhang Y. Transcriptomics-Based Identification of Genes Related to Tapetum Degradation and Microspore Development in Lily. Genes. 2022; 13(2):366. https://doi.org/10.3390/genes13020366
Chicago/Turabian StyleSui, Juanjuan, Wenjie Jia, Yin Xin, and Yuanyuan Zhang. 2022. "Transcriptomics-Based Identification of Genes Related to Tapetum Degradation and Microspore Development in Lily" Genes 13, no. 2: 366. https://doi.org/10.3390/genes13020366
APA StyleSui, J., Jia, W., Xin, Y., & Zhang, Y. (2022). Transcriptomics-Based Identification of Genes Related to Tapetum Degradation and Microspore Development in Lily. Genes, 13(2), 366. https://doi.org/10.3390/genes13020366