
Analysing miRNA-Target Gene Networks in Inflammatory Bowel Disease and Other Complex Diseases Using Transcriptomic Data

 , and
, and
Abstract
:1. Introduction
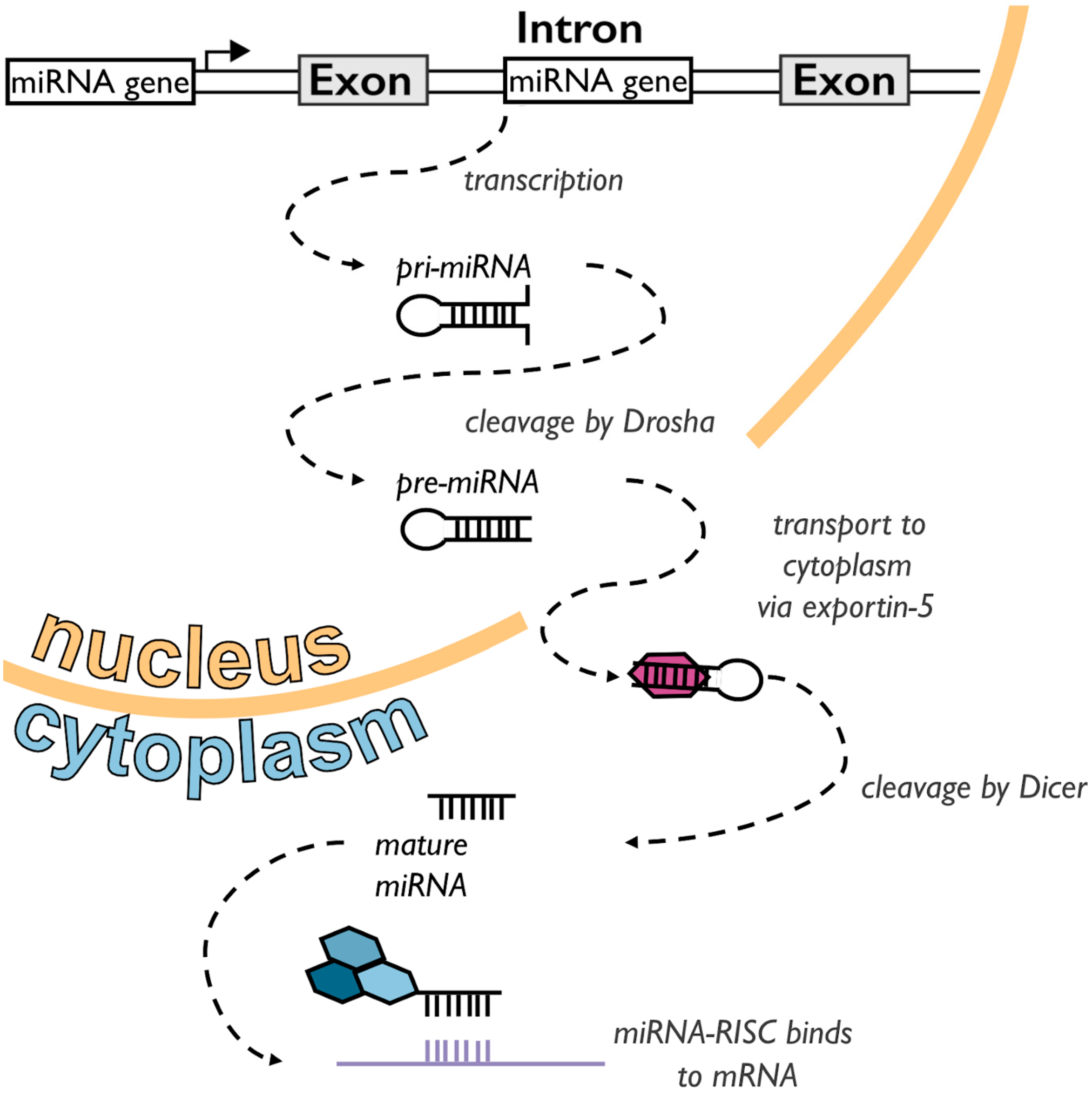
1.1. A Short Primer on miRNAs
1.2. miRNAs in IBD
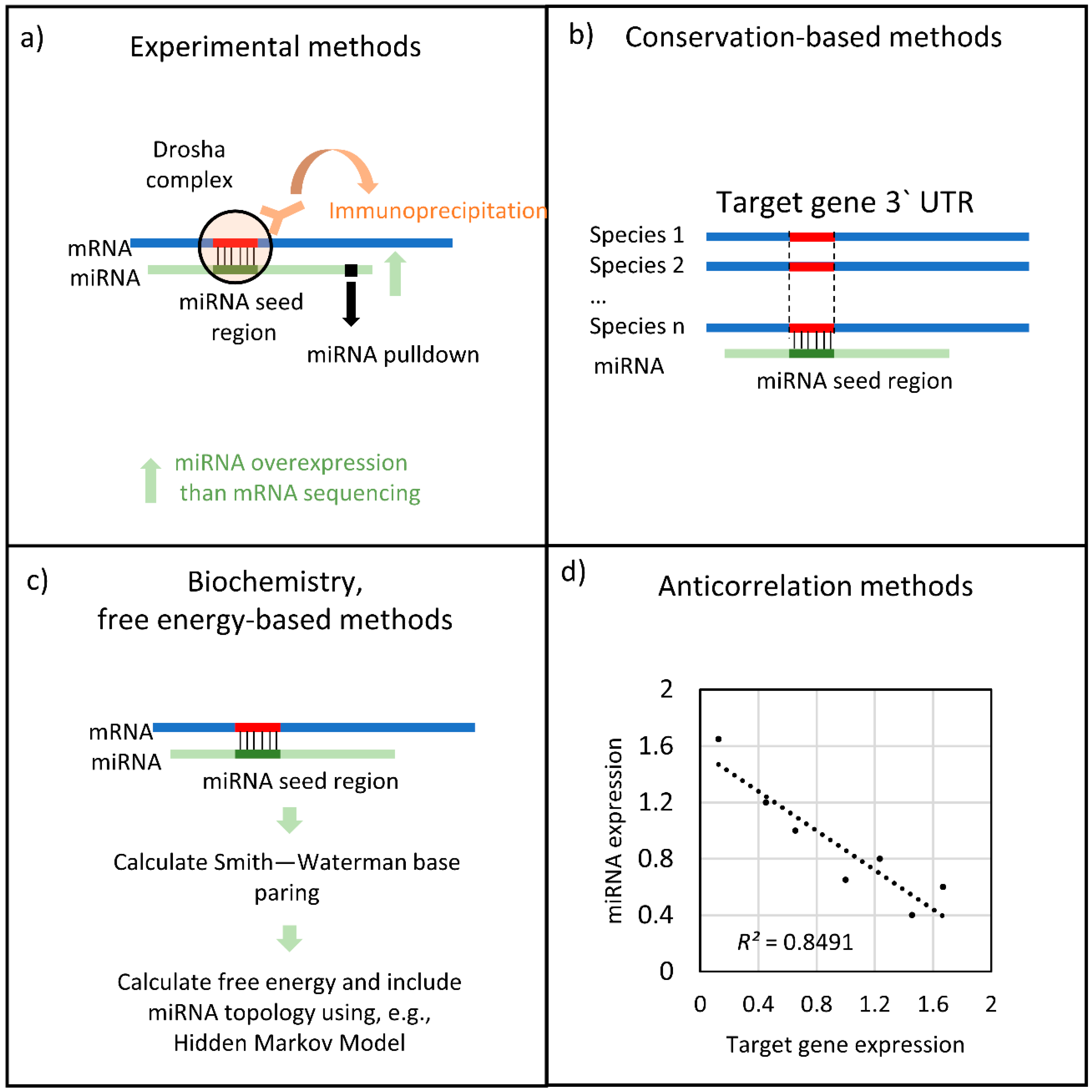
2. Bioinformatics Approaches for Predicting miRNA Function in Complex Disease
2.1. Similarity-Based Methodologies
2.2. Transcription-Based Methodologies
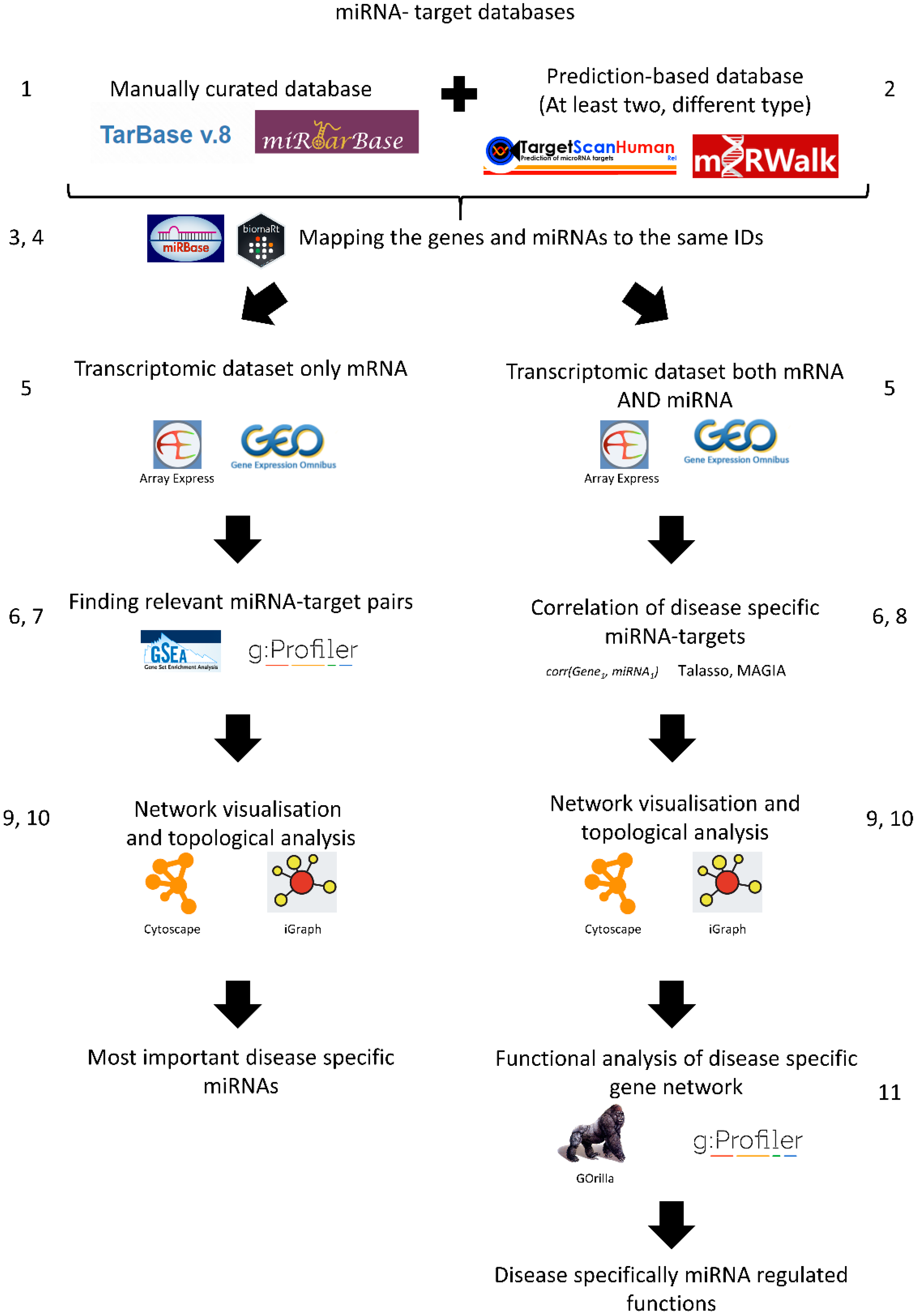
3. Materials and Methods: Protocol for Transcriptome-Based miRNA-Target Gene Analysis for Complex Disorders
- Strengthening predictions using additional miRNA–mRNA target databases: Manually curated databases do not have all miRNA-target gene interactions. Hence, it is useful to add an additional miRNA-target gene interaction database which contains predicted miRNA-target gene interactions. The best approach is to use multiple data sources from at least two complementary methods, e.g., TargetScan and miranda/mirSVR.
- Combining the miRNA-target gene networks using the same miRNA IDs: Unfortunately, miRNAs are not always consistently named between databases. miRBase is the de facto database for miRNA families and sequences and contains a basic ID mapping tool [28,29]. MiRBase has an R package for application programming interface (API) access called miRBaseConverter [30]. Please note, in the case of TargetScan, the miRNA family-based conversions can cause many-to-many mapping issues.
- Combining the miRNA-target gene networks using the same target gene IDs: The same problem can arise when mapping target genes. A good solution is to use the biomaRt R package, or download the various annotation tables from UniProt [31] or Ensembl biomart [32]. It is important to choose one type of gene ID when constructing the gene network.
- Preparing gene expression data: The transcriptomics data can be downloaded with normalised log2 counts or expression values from Gene Expression Omnibus (GEO) [33], ArrayExpress [34], or analysed in-house. Ideally, both mRNA and miRNA transcriptome data are available, but often this is not the case. In the case of RNA-seq data analysis, we suggest using the normalised count table with genes considered as expressed if they have more than 1 count per million. However, the exact normalisation between the samples and the filtering of weakly expressed genes are choices the researcher should make depending on the sequencing depth, read quality, and read length [35,36].
- Finding differentially expressed genes: From the normalised counts, the next step is to calculate differentially expressed genes (DEGs), e.g., using the limma package from R. Limma uses the moderated t-statistic to calculate differential gene expression [37]. We recommend |log2FC| > 1 and Benjamini–Hochberg (BH) corrected p value < 0.05, but again it can change depending on the researcher’s questions and quality of samples. It is important that these results are corrected for false discovery rate and filtered by fold change as well.
- Enrichment of potential miRNA targets in the differentially expressed genes (if miRNA expression values are not available): If the miRNA expression values are not available, it is possible to enrich potential regulatory miRNAs using the constructed miRNA gene network. To do this, the used gene network needs to be transformed as a gene matrix (GMT) file. The constructed GMT file can be used as an input of a gene set enrichment analysis (GSEA) [38]. In this case, the expression values are used for calculations. Moreover, the DEGs with the GMT file can be used for enrichment analysis, for instance using gprofiler2 [39]. It is important that the enrichment background, the intersection of miRNA-regulated genes, and the measured transcripts are utilised.
- Building an anticorrelation network (if the miRNA expression data are available): If miRNA expression data are available, then the prediction of miRNA and target genes can be calculated, e.g., from the TaLasso algorithm [40] which has an R package available or the miRLAB package [41]. The correlation-based network needs to be filtered with the previously generated miRNA-target gene network to identify the dataset-specific miRNA-target gene interactions. The MAGIA2 method can be used for steps 2–8 to produce the output networks [42]. We suggest using high correlation values such as the 25th percentile of the most negatively correlating miRNAs.
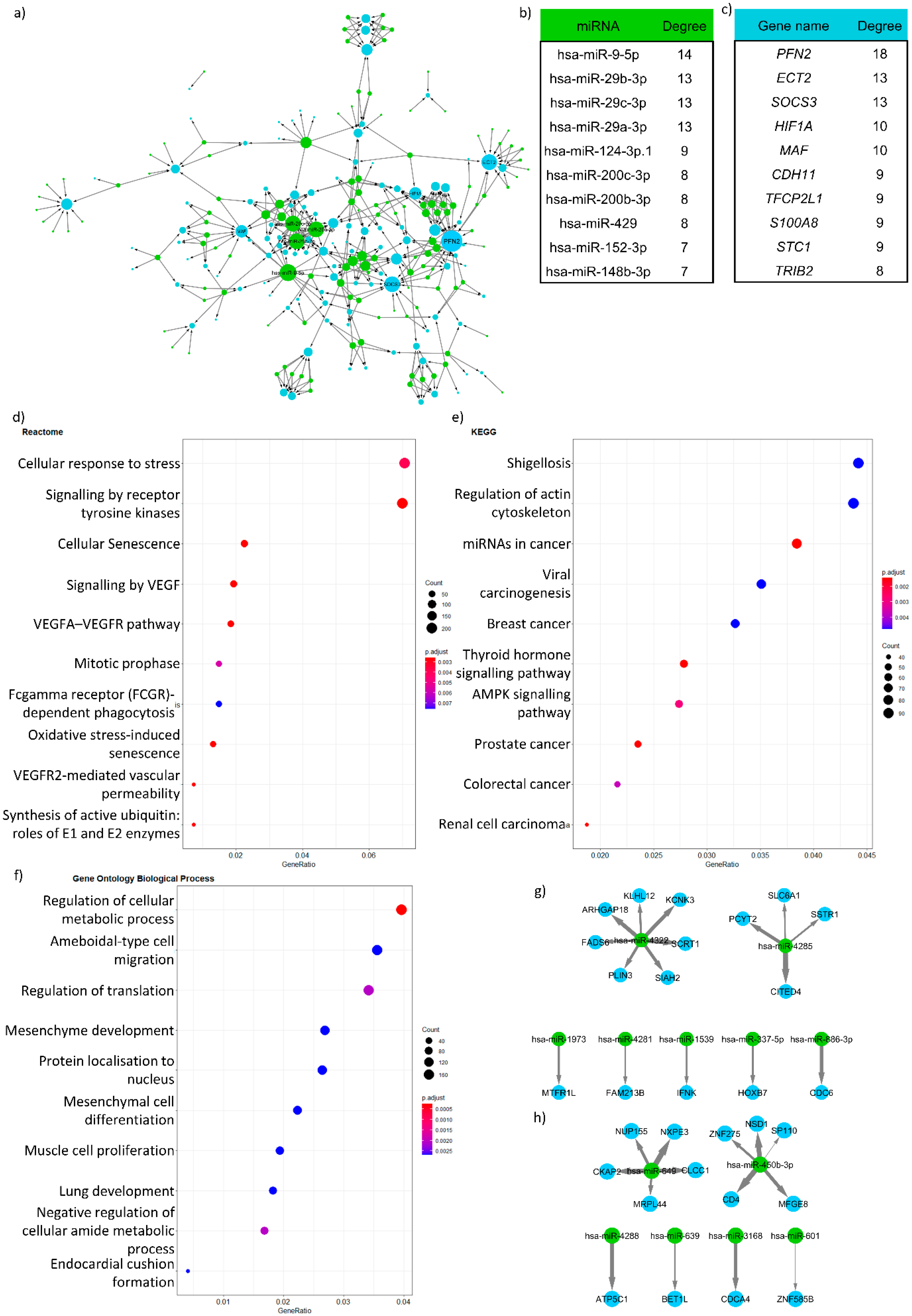
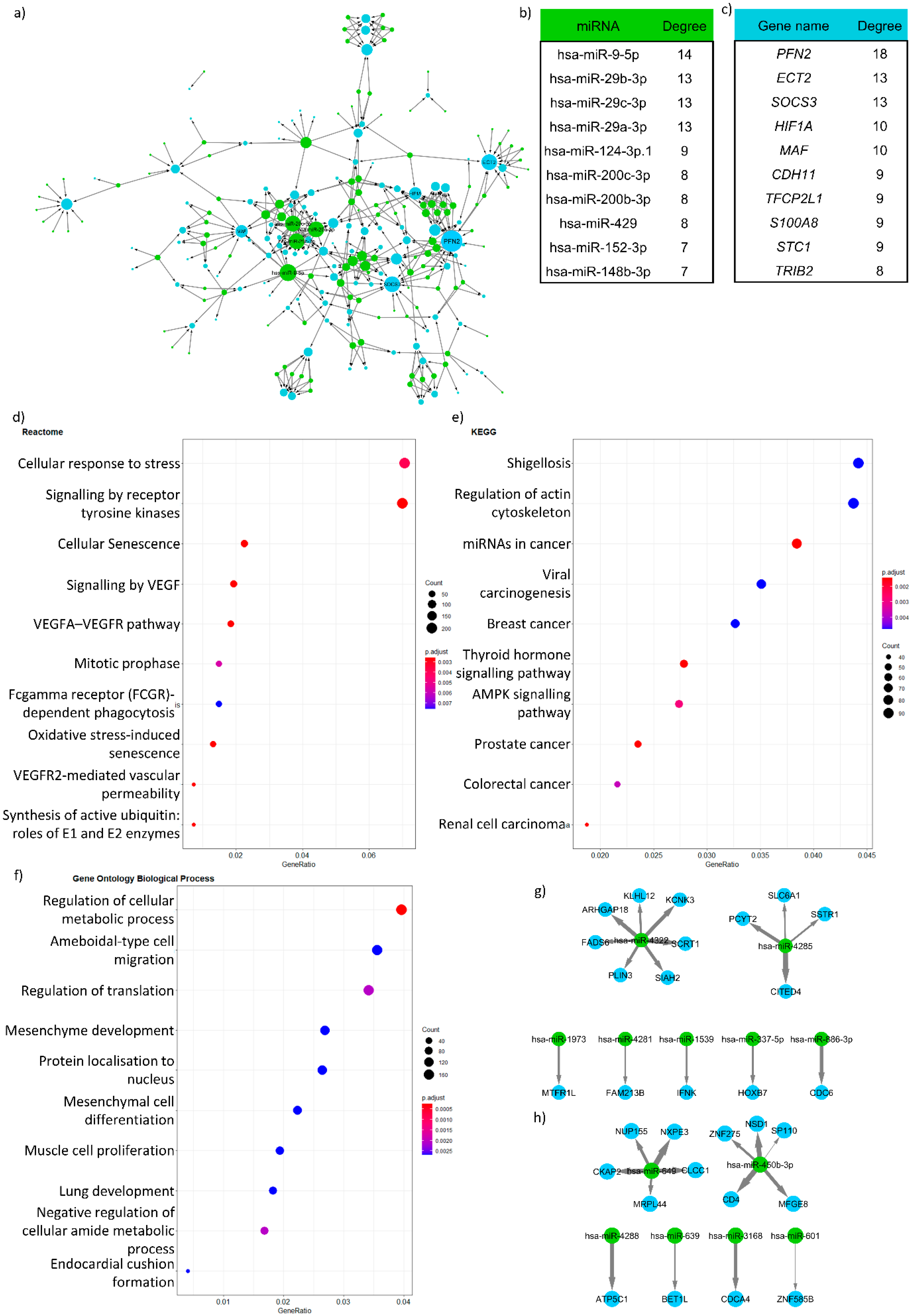
- Visualising the networks: After creating the networks where the potential miRNA targets are given, the next step is to visualise the network using network visualisation tools. The most commonly used visualisation tool in network biology is Cytoscape [43]. If programmatic access is required, the R and python software package igraph can be used [44].
- Analysing the resulting visualised network: The most common way to analyse the visualised network is by looking at the hubs in the network, i.e., nodes that have a high number of edges or degree. Those target genes or miRNAs with a high degree (“hub” genes/miRNAs) have a greater chance to be involved in the disease of interest (in this case IBD). For identifying these nodes, the Cytoscape network analyser can be used. We suggest that the top ten percent of highest degree nodes can be considered as hub genes, but this also depends on the degree distribution of the network.
- Functional analysis: The next step is to evaluate what functions of the network are potentially regulated by miRNAs compared to the whole network. This can be performed by gene ontology enrichment analysis, e.g., the aforementioned gprofiler2 [37] package or the GOrilla analysis tool [45]. An important caveat to bear in mind is that miRNA target genes are limited by the used miRNA-target gene network. Therefore, the background of any enrichment analysis needs to be modified specifically. In the background, only the genes in the miRNA-target gene network should be utilised. If the differentially expressed genes are directly used for building the miRNA-target gene network, then it is advisable not to perform enrichment analysis because this would, in effect, simply test differentially expressed genes again.
- Experimental validation: The next step would be the experimental validation of the detected miRNA and target gene anti-correlation, which is beyond the scope of this technical note.
4. Results—Case Study in Crohn’s Disease
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roda, G.; Ng, S.C.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Prim. 2020, 6, 22. [Google Scholar] [CrossRef]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Prim. 2020, 6, 74. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Laval, G.; Quach, H.; Patin, E.; Quintana-Murci, L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 2008, 40, 340–345. [Google Scholar] [CrossRef]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-K.; Kim, V.N. Processing of intronic microRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Merritt, W.M.; Bar-Eli, M.; Sood, A.K. The dicey role of Dicer: Implications for RNAi therapy. Cancer Res. 2010, 70, 2571–2574. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microrna biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, S.; Purohit, P.; Sharma, P. Micrornas and diseases: Promising biomarkers for diagnosis and therapeutics. Indian J. Clin. Biochem. 2019, 34, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Soroosh, A.; Koutsioumpa, M.; Pothoulakis, C.; Iliopoulos, D. Functional role and therapeutic targeting of microRNAs in inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G256–G262. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, S.; De Hertogh, G.; Van der Goten, J.; Verstockt, B.; Vancamelbeke, M.; Machiels, K.; Van Lommel, L.; Schuit, F.; Van Assche, G.; Rutgeerts, P.; et al. Gene and mirna regulatory networks during different stages of crohn’s disease. J. Crohns Colitis 2019, 13, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Viennois, E.; Zhao, Y.; Han, M.K.; Xiao, B.; Zhang, M.; Prasad, M.; Wang, L.; Merlin, D. Serum miRNA signature diagnoses and discriminates murine colitis subtypes and predicts ulcerative colitis in humans. Sci. Rep. 2017, 7, 2520. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Guo, N.J.; Tian, H.; Marohn, M.; Gearhart, S.; Bayless, T.M.; Brant, S.R.; Kwon, J.H. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Xie, Y.; Fu, Y.; Zhou, Q.; Ma, Z.; Ma, J.; Huang, Z.; Zhang, J.; Chen, J. The signaling axis of microRNA-31/interleukin-25 regulates Th1/Th17-mediated inflammation response in colitis. Mucosal Immunol. 2017, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Sumathipala, M.; Weiss, S.T. Predicting miRNA-based disease-disease relationships through network diffusion on multi-omics biological data. Sci. Rep. 2020, 10, 8705. [Google Scholar] [CrossRef] [PubMed]
- Mørk, S.; Pletscher-Frankild, S.; Caro, A.P.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA-disease associations. Bioinformatics 2014, 30, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, Q.-F.; Yan, G.-Y. RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction. RNA Biol. 2017, 14, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Ping, Y.; Li, X.; Zhao, H.; Wang, L.; Fan, H.; Xiao, Y.; Li, X. Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol. Biosyst. 2014, 10, 2800–2809. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.P.; Modos, D.; Korcsmaros, T.; Brooks-Warburton, J. Network biology approaches to achieve precision medicine in inflammatory bowel disease. Front. Genet. 2021, 12, 760501. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Su, N.; Liu, L.; Zhang, J.; Wang, H.; Zhang, W.; Gui, J.; Yu, K.; Li, J.; Le, T.D. miRBaseConverter: An R/Bioconductor package for converting and retrieving miRNA name, accession, sequence and family information in different versions of miRBase. BMC Bioinform. 2018, 19, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt Consortium UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [CrossRef] [PubMed]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazma, A.; Parkinson, H.; Sarkans, U.; Shojatalab, M.; Vilo, J.; Abeygunawardena, N.; Holloway, E.; Kapushesky, M.; Kemmeren, P.; Lara, G.G.; et al. ArrayExpress—A public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003, 31, 68–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, 709. [Google Scholar] [CrossRef] [PubMed]
- Muniategui, A.; Nogales-Cadenas, R.; Vázquez, M.; Aranguren, X.L.; Agirre, X.; Luttun, A.; Prosper, F.; Pascual-Montano, A.; Rubio, A. Quantification of miRNA-mRNA interactions. PLoS ONE 2012, 7, e30766. [Google Scholar] [CrossRef]
- Le, T.D.; Zhang, J.; Liu, L.; Liu, H.; Li, J. miRLAB: An R Based Dry Lab for Exploring miRNA-mRNA Regulatory Relationships. PLoS ONE 2015, 10, e0145386. [Google Scholar] [CrossRef] [Green Version]
- Bisognin, A.; Sales, G.; Coppe, A.; Bortoluzzi, S.; Romualdi, C. MAGIA2: From miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update). Nucleic Acids Res. 2012, 40, W13–W21. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.-D.; Lin, F.-M.; Wu, W.-Y.; Liang, C.; Huang, W.-C.; Chan, W.-L.; Tsai, W.-T.; Chen, G.-Z.; Lee, C.-J.; Chiu, C.-M.; et al. miRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.-D.; Tseng, Y.-T.; Shrestha, S.; Lin, Y.-L.; Khaleel, A.; Chou, C.-H.; Chu, C.-F.; Huang, H.-Y.; Lin, C.-M.; Ho, S.-Y.; et al. miRTarBase update 2014: An information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014, 42, D78–D85. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, Y.H. miRDeathDB: A database bridging microRNAs and the programmed cell death. Cell Death Differ. 2012, 19, 1571. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [Green Version]
- Volinia, S.; Visone, R.; Galasso, M.; Rossi, E.; Croce, C.M. Identification of microRNA activity by Targets’ Reverse EXpression. Bioinformatics 2010, 26, 91–97. [Google Scholar] [CrossRef]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yue, D.; Chen, Y.; Gao, S.-J.; Huang, Y. Improving performance of mammalian microRNA target prediction. BMC Bioinform. 2010, 11, 476. [Google Scholar] [CrossRef] [Green Version]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Gennarino, V.A.; Sardiello, M.; Mutarelli, M.; Dharmalingam, G.; Maselli, V.; Lago, G.; Banfi, S. HOCTAR database: A unique resource for microRNA target prediction. Gene 2011, 480, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Hecker, N.; Stephan, C.; Mollenkopf, H.-J.; Jung, K.; Preissner, R.; Meyer, H.-A. A new algorithm for integrated analysis of miRNA-mRNA interactions based on individual classification reveals insights into bladder cancer. PLoS ONE 2013, 8, e64543. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Quillet, A.; Saad, C.; Ferry, G.; Anouar, Y.; Vergne, N.; Lecroq, T.; Dubessy, C. Improving Bioinformatics Prediction of microRNA Targets by Ranks Aggregation. Front. Genet. 2019, 10, 1330. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, Y. Current experimental strategies for intracellular target identification of microRNA. ExRNA 2019, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.C.; Babak, T.; Corson, T.W.; Chua, G.; Khan, S.; Gallie, B.L.; Hughes, T.R.; Blencowe, B.J.; Frey, B.J.; Morris, Q.D. Using expression profiling data to identify human microRNA targets. Nat. Methods 2007, 4, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Maki, M.; Ding, R.; Yang, Y.; Zhang, B.; Xiong, L. Genome-wide survey of tissue-specific microRNA and transcription factor regulatory networks in 12 tissues. Sci. Rep. 2014, 4, 5150. [Google Scholar] [CrossRef] [Green Version]
- Sood, P.; Krek, A.; Zavolan, M.; Macino, G.; Rajewsky, N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. USA 2006, 103, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Farh, K.K.-H.; Grimson, A.; Jan, C.; Lewis, B.P.; Johnston, W.K.; Lim, L.P.; Burge, C.B.; Bartel, D.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science 2005, 310, 1817–1821. [Google Scholar] [CrossRef] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Name | Description | Website (Access Date) | Reference |
|---|---|---|---|---|
| General | Mirbase | The de facto miRNA central repository for miRNA families and sequences | http://www.mirbase.org (16 January 2022) | [28] |
| Literature curation | miRTarBase | Experimentally proven miRNA-target gene connections | http://mirtarbase.mbc.nctu.edu.cn (16 January 2022) | [46,47] |
| Literature curation | TarBase | Experimentally validated miRNA-target gene connections | http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex (16 January 2022) | [26] |
| Literature curation | miRDeathDB | Small database of experimentally validated miRNA-target interactions related to cell death | http://www.rna-world.org/mirdeathdb (25 April 2016) | [48] |
| Literature curation | miR2Disease | Small literature curation database for miRNAs associated with diseases | http://www.mir2disease.org (16 January 2022) | [49] |
| Conservation based | Targetscan | The largest miRNA-target gene prediction database based on sequence homology | http://www.targetscan.org/ (16 January 2022) | [50] |
| Conservation based | PicTar | Old sequence homology-based miRNA-target database | http://pictar.mdc-berlin.de (16 January 2022) | [51] |
| Biochemistry based | Miranda | A free energy-based algorithm for miRNA-target prediction currently unavailable, but still widely used | http://www.microrna.org/microrna/home.do (16 June 2020) | [52] |
| Biochemistry and conservation based | SVMicrO | Two-stage support vector machine-based miRNA-target prediction algorithm and database integrating biochemistry, alignment, and conversation features of the target site and the miRNA | http://compgenomics.utsa.edu/svmicro.html (16 January 2022) | [53] |
| Biochemistry based | PITA | Thermodynamics-based prediction tool which incorporates the target’s accessibility | https://genie.weizmann.ac.il/pubs/mir07/mir07_prediction.html (16 January 2022) | [54] |
| Expression based | hoctar | It uses various prediction tools and then multiple miRNA and target gene expression datasets to calculate the potential miRNA-target gene connections | https://hoctar.tigem.it/ (16 January 2022) | [55] |
| Expression based | CAPE RNA | miRNA-target gene prediction tools using discrete mRNA and miRNA levels (middle, high, low) | https://sourceforge.net/projects/caperna (16 January 2022) | [56] |
| Literature curation and predicted targets | miRecords | Joint effort of multiple prediction tools and validated targets. Last updated in 2013. | http://c1.accurascience.com/miRecords (16 January 2022) | [57] |
| Large collection of multiple different resources | mirwalk | Contains 13 different target prediction methods and it generates predictions for the whole length of the genes | http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2 (16 January 2022) | [58] |
| Large integrated database | miRabel | Contains integrated predictions from multiple prediction databases and also the experimentally validated informations of miRecords and miRTarBase | http://bioinfo.univ-rouen.fr/mirabel (16 January 2022) | [59] |
| Integrated tool for expression-based prediction | MAGIA | Prediction tool using miRNA and mRNA gene expression to integrate with various miRNA-target prediction algorithms | http://gencomp.bio.unipd.it/magia2/start (25 August 2018) | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.P.; Ölbei, M.; Brooks-Warburton, J.; Korcsmaros, T.; Modos, D. Analysing miRNA-Target Gene Networks in Inflammatory Bowel Disease and Other Complex Diseases Using Transcriptomic Data. Genes 2022, 13, 370. https://doi.org/10.3390/genes13020370
Thomas JP, Ölbei M, Brooks-Warburton J, Korcsmaros T, Modos D. Analysing miRNA-Target Gene Networks in Inflammatory Bowel Disease and Other Complex Diseases Using Transcriptomic Data. Genes. 2022; 13(2):370. https://doi.org/10.3390/genes13020370
Chicago/Turabian StyleThomas, John P., Marton Ölbei, Johanne Brooks-Warburton, Tamas Korcsmaros, and Dezso Modos. 2022. "Analysing miRNA-Target Gene Networks in Inflammatory Bowel Disease and Other Complex Diseases Using Transcriptomic Data" Genes 13, no. 2: 370. https://doi.org/10.3390/genes13020370
APA StyleThomas, J. P., Ölbei, M., Brooks-Warburton, J., Korcsmaros, T., & Modos, D. (2022). Analysing miRNA-Target Gene Networks in Inflammatory Bowel Disease and Other Complex Diseases Using Transcriptomic Data. Genes, 13(2), 370. https://doi.org/10.3390/genes13020370