
Does the Expression and Epigenetics of Genes Involved in Monogenic Forms of Parkinson’s Disease Influence Sporadic Forms?

 , ,
, ,
Abstract
:1. Introduction
2. α Synuclein
2.1. Function and Subcellular Distribution of α-Syn in PD
2.2. Overexpression of α-Syn in PD
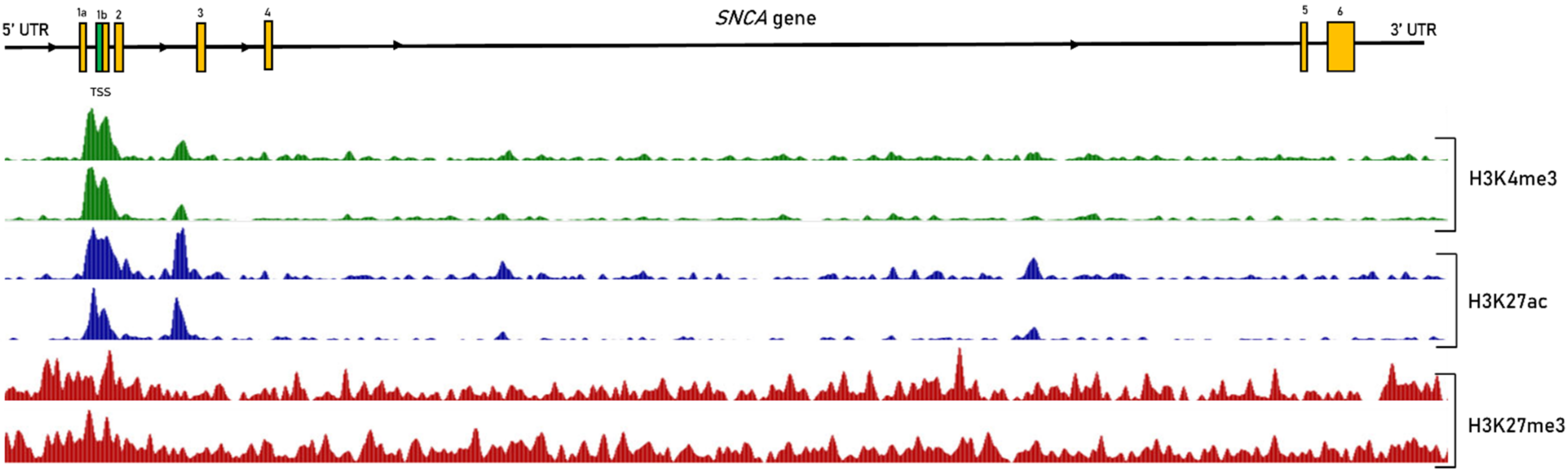
2.3. Epigenetic Regulation of SNCA Expression in Sporadic PD
2.4. Non-Coding RNAs Regulating SNCA Transcription
2.5. Interplay between Epigenetic Mechanisms in the Regulation of SNCA
3. LRRK2
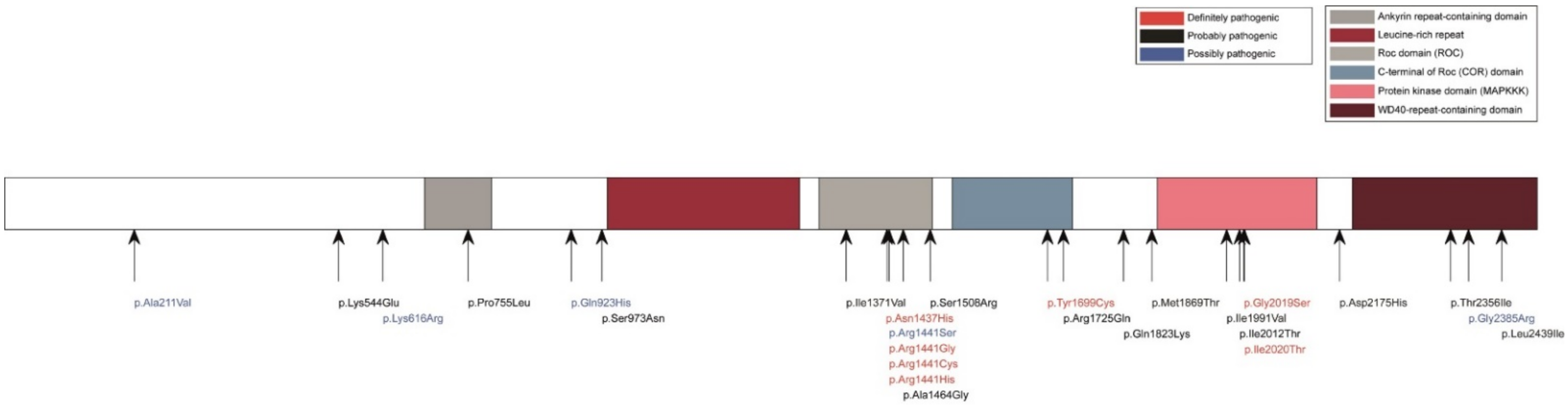
3.1. LRRK2 Protein Function and Localization
3.2. Regulation of LRRK2 Transcription
3.3. LRRK2 in Immune Cells
4. Dysregulation of Genes Involved in Recessive Forms of PD
4.1. Expression Profile of PRKN and Its Regulation
4.2. Role of Parkin and Its Epigenetic Regulation in Sporadic PD
4.3. PINK1 Expression in PD Patient Brains
4.4. Regulation of DJ-1 Expression
5. GBA a Risk Factor for PD
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s Disease: Genetics and Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 193–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Goll, M.G.; Bestor, T.H. Eukaryotic Cytosine Methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Zhuo, Y.; Shan, B. MicroRNAs, long noncoding RNAs, and their functions in human disease. In Bioinformatics in MicroRNA Research; Huang, J., Borchert, G.M., Dou, D., Huan, J., Lan, W., Tan, M., Wu, B., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1617, pp. 1–25. [Google Scholar] [CrossRef]
- Argonaute Proteins: Functional Insights and Emerging Roles|Nature Reviews Genetics. Available online: https://www.nature.com/articles/nrg3462 (accessed on 26 October 2021).
- miRNA Activity Inferred from Single Cell mRNA Expression|Scientific Reports. Available online: https://www.nature.com/articles/s41598-021-88480-5 (accessed on 24 November 2021).
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef]
- Gründemann, J.; Schlaudraff, F.; Haeckel, O.; Liss, B. Elevated α-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res. 2008, 36, e38. [Google Scholar] [CrossRef] [Green Version]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation Regulates α-Synuclein Expression and Is Decreased in Parkinson’s Disease Patients’ Brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG Demethylation Enhances α-Synuclein Expression and Affects the Pathogenesis of Parkinson’s Disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [Green Version]
- Pihlstrøm, L.; Berge, V.; Rengmark, A.; Toft, M. Parkinson’s disease correlates with promoter methylation in the α-synuclein gene. Mov. Disord. 2015, 30, 577–580. [Google Scholar] [CrossRef]
- Ai, S.-X.; Xu, Q.; Hu, Y.-C.; Song, C.-Y.; Guo, J.-F.; Shen, L.; Wang, C.-R.; Yu, R.-L.; Yan, X.-X.; Tang, B.-S. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J. Neurol. Sci. 2014, 337, 123–128. [Google Scholar] [CrossRef]
- Tan, Y.-Y.; Wu, L.; Zhao, Z.-B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.-F.; Chen, S.-D. Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Park. Relat. Disord. 2014, 20, 308–313. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.A.; Kannarkat, G.T.; Cintron, A.F.; Butkovich, L.M.; Fraser, K.B.; Chang, J.; Grigoryan, N.; Factor, S.A.; West, A.B.; Boss, J.M.; et al. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinson’s Dis. 2017, 3, 11. [Google Scholar] [CrossRef]
- Beyer, K.; Domingo-Sàbat, M.; Humbert, J.; Carrato, C.; Ferrer, I.; Ariza, A. Differential expression of α-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 2008, 9, 163–172. [Google Scholar] [CrossRef]
- Cai, M.; Tian, J.; Zhao, G.-H.; Luo, W.; Zhang, B.-R. Study of Methylation Levels of Parkin Gene Promoter in Parkinson’s Disease Patients. Int. J. Neurosci. 2011, 121, 497–502. [Google Scholar] [CrossRef]
- De Mena, L.; Cardo, L.F.; Coto, E.; Alvarez, V.; Coto, E. No differential DNA methylation of PARK2 in brain of Parkinson’s disease patients and healthy controls. Mov. Disord. 2013, 28, 2032–2033. [Google Scholar] [CrossRef]
- Eryilmaz, I.E.; Cecener, G.; Erer, S.; Egeli, U.; Tunca, B.; Zarifoglu, M.; Elibol, B.; Tokcaer, A.B.; Saka, E.; Demirkiran, M.; et al. Epigenetic approach to early-onset Parkinson’s disease: Low methylation status of SNCA and PARK2 promoter regions. Neurol. Res. 2017, 39, 965–972. [Google Scholar] [CrossRef]
- Ding, H.; Huang, Z.; Chen, M.; Wang, C.; Chen, X.; Chen, J.; Zhang, J. Identification of a panel of five serum miRNAs as a biomarker for Parkinson’s disease. Park. Relat. Disord. 2016, 22, 68–73. [Google Scholar] [CrossRef]
- Xing, R.-X.; Li, L.-G.; Liu, X.-W.; Tian, B.-X.; Cheng, Y. Down regulation of miR-218, miR-124, and miR-144 relates to Parkinson’s disease via activating NF-κB signaling. Kaohsiung J. Med. Sci. 2020, 36, 786–792. [Google Scholar] [CrossRef]
- Serafin, A.; Foco, L.; Zanigni, S.; Blankenburg, H.; Picard, A.; Zanon, A.; Giannini, G.; Pichler, I.; Facheris, M.F.; Cortelli, P.; et al. Overexpression of blood microRNAs 103a, 30b, and 29a in L-dopa-treated patients with PD. Neurology 2015, 84, 645–653. [Google Scholar] [CrossRef]
- Muqit, M.M.K.; Abou-Sleiman, P.M.; Saurin, A.T.; Harvey, K.; Gandhi, S.; Deas, E.; Eaton, S.; Smith, M.D.P.; Venner, K.; Matilla, A.; et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J. Neurochem. 2006, 98, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Blackinton, J.G.; Anvret, A.; Beilina, A.; Olson, L.; Cookson, M.R.; Galter, D. Expression of PINK1 mRNA in human and rodent brain and in Parkinson’s disease. Brain Res. 2007, 1184, 10–16. [Google Scholar] [CrossRef]
- Navarro-Sánchez, L.; Águeda-Gómez, B.; Aparicio, S.; Pérez-Tur, J. Epigenetic Study in Parkinson’s Disease: A Pilot Analysis of DNA Methylation in Candidate Genes in Brain. Cells 2018, 7, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazeli, S.; Motovali-Bashi, M.; Peymani, M.; Hashemi, M.-S.; Etemadifar, M.; Nasr-Esfahani, M.H.; Ghaedi, K. A compound downregulation of SRRM2 and miR-27a-3p with upregulation of miR-27b-3p in PBMCs of Parkinson’s patients is associated with the early stage onset of disease. PLoS ONE 2020, 15, e0240855. [Google Scholar]
- Dos Santos, M.C.T.; Barreto-Sanz, M.A.; Correia, B.R.S.; Bell, R.; Widnall, C.; Perez, L.T.; Berteau, C.; Schulte, C.; Scheller, D.; Berg, D.; et al. miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget 2018, 9, 17455–17465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaran, R.; Vandrovcova, J.; Luk, C.; Sharma, S.; Renton, A.; Wood, N.; Hardy, J.A.; Lees, A.J.; Bandopadhyay, R. Differential DJ-1 gene expression in Parkinson’s disease. Neurobiol. Dis. 2009, 36, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wu, L.; Li, D.; Liu, X.; Ding, J.; Chen, S. Methylation status of DJ-1 in leukocyte DNA of Parkinson’s disease patients. Transl. Neurodegener. 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Gao, C.; Sun, Q.; Pan, H.; Huang, P.; Ding, J.; Chen, S. MicroRNA-4639 Is a Regulator of DJ-1 Expression and a Potential Early Diagnostic Marker for Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [Green Version]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Kalia, S.K.; McLean, P.; Lozano, A.M.; Lang, A.E. α-Synuclein oligomers and clinical implications for Parkinson disease. Ann. Neurol. 2012, 73, 155–169. [Google Scholar] [CrossRef]
- Erskine, D.; Patterson, L.; Alexandris, A.; Hanson, P.S.; McKeith, I.G.; Attems, J.; Morris, C.M. Regional levels of physiological α-synuclein are directly associated with Lewy body pathology. Acta Neuropathol. 2018, 135, 153–154. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.R.; Rhoades, E. Effects of Curvature and Composition on α-Synuclein Binding to Lipid Vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [Green Version]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. α-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [Green Version]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 Inhibitors Rescue α-Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Mutez, E.; Lepretre, F.; Le Rhun, E.; Larvor, L.; Duflot, A.; Mouroux, V.; Kerckaert, J.; Figeac, M.; Dujardin, K.; Destée, A.; et al. SNCA locus duplication carriers: From genetics to Parkinson disease phenotypes. Hum. Mutat. 2011, 32, E2079–E2090. [Google Scholar] [CrossRef] [Green Version]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of α-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Chiba-Falek, O.; Lopez, G.J.; Nussbaum, R.L. Levels of α-synuclein mRNA in sporadic Parkinson disease patients. Mov. Disord. 2006, 21, 1703–1708. [Google Scholar] [CrossRef]
- McLean, J.R.; Hallett, P.J.; Cooper, O.; Stanley, M.; Isacson, O. Transcript expression levels of full-length α-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of α-synuclein overexpression. Mol. Cell. Neurosci. 2012, 49, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Wong, H.; Guttman, M.; Ang, L.C.; Forno, L.S.; Shimadzu, M.; Rajput, A.H.; Muenter, M.D.; Kish, S.J.; Hornykiewicz, O.; et al. Brain α-synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: A comparative investigation. Brain 2010, 133, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein Sequesters Dnmt1 from the Nucleus. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Li, J.; Zhang, Z.; Wang, H.; Wang, Z. Epigenetic upregulation of α-synuclein in the rats exposed to methamphetamine. Eur. J. Pharmacol. 2014, 745, 243–248. [Google Scholar] [CrossRef]
- Schmitt, I.; Kaut, O.; Khazneh, H.; Deboni, L.; Ahmad, A.; Berg, D.; Klein, C.; Fröhlich, H.; Wüllner, U. L-dopa increases α-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov. Disord. 2015, 30, 1794–1801. [Google Scholar] [CrossRef]
- De Lau, L.M.L.; Breteler, M.M.B. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y.-S. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 2021, 13, e12188. [Google Scholar] [CrossRef]
- Vermunt, M.W.; Reinink, P.; Korving, J.; De Bruijn, E.; Creyghton, P.M.; Basak, O.; Geeven, G.; Toonen, P.W.; Lansu, N.; Meunier, C.; et al. Large-Scale Identification of Coregulated Enhancer Networks in the Adult Human Brain. Cell Rep. 2014, 9, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voutsinas, G.E.; Stavrou, E.F.; Karousos, G.; Dasoula, A.; Papachatzopoulou, A.; Syrrou, M.; Verkerk, A.J.; Van der Spek, P.; Patrinos, G.P.; Stöger, R.; et al. Allelic imbalance of expression and epigenetic regulation within the α-synuclein wild-type and p.Ala53Thr alleles in Parkinson disease. Hum. Mutat. 2010, 31, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, S.; Bjørnevik, K.; Soon Im, D.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a Regulator of the α-Synuclein Gene Driving Risk of Parkinson’s Disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear α-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of α-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [Green Version]
- Doxakis, E. Post-transcriptional Regulation of α-Synuclein Expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [Green Version]
- Fragkouli, A.; Doxakis, E. miR-7 and miR-153 protect neurons against MPP+-induced cell death via upregulation of mTOR pathway. Front. Cell. Neurosci. 2014, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.C.; Yoo, M.; Kabaria, S.; Junn, E. MicroRNA-7 facilitates the degradation of α-synuclein and its aggregates by promoting autophagy. Neurosci. Lett. 2018, 678, 118–123. [Google Scholar] [CrossRef]
- Villar-Menéndez, I.; Porta, S.; Buira, S.P.; Pereira-Veiga, T.; Díaz-Sánchez, S.; Albasanz, J.L.; Ferrer, I.; Martín, M.; Barrachina, M. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol. Dis. 2014, 69, 206–214. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett. 2014, 589, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Rhinn, H.; Qiang, L.; Yamashita, T.; Rhee, D.; Zolin, A.; Vanti, W.; Abeliovich, A. α-Synuclein transcript alternative 3′UTR usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 2012, 3, 1084. [Google Scholar] [CrossRef]
- Consales, C.; Cirotti, C.; Filomeni, G.; Panatta, M.; Butera, A.; Merla, C.; Lopresto, V.; Pinto, R.; Marino, C.; Benassi, B. Fifty-Hertz Magnetic Field Affects the Epigenetic Modulation of the miR-34b/c in Neuronal Cells. Mol. Neurobiol. 2018, 55, 5698–5714. [Google Scholar] [CrossRef]
- Mizuta, I.; Takafuji, K.; Ando, Y.; Satake, W.; Kanagawa, M.; Kobayashi, K.; Nagamori, S.; Shinohara, T.; Ito, C.; Yamamoto, M.; et al. YY1 binds to α-synuclein 3′-flanking region SNP and stimulates antisense noncoding RNA expression. J. Hum. Genet. 2013, 58, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Segal, T.; Salmon-Divon, M.; Gerlitz, G. The Heterochromatin Landscape in Migrating Cells and the Importance of H3K27me3 for Associated Transcriptome Alterations. Cells 2018, 7, 205. [Google Scholar] [CrossRef] [Green Version]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Hughes, A.L.; Kelley, J.R.; Klose, R.J. Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194567. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Marchand, A.; Drouyer, M.; Sarchione, A.; Chartier-Harlin, M.-C.; Taymans, J.-M. LRRK2 Phosphorylation, More Than an Epiphenomenon. Front. Neurosci. 2020, 14, 527. [Google Scholar] [CrossRef]
- Deyaert, E.; Wauters, L.; Guaitoli, G.; Konijnenberg, A.; Leemans, M.; Terheyden, S.; Petrovic, A.; Gallardo, R.; Nederveen-Schippers, L.M.; Athanasopoulos, P.; et al. A homologue of the Parkinson’s disease-associated protein LRRK2 undergoes a monomer-dimer transition during GTP turnover. Nat. Commun. 2017, 8, 1008. [Google Scholar] [CrossRef]
- Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. LRRK2, a puzzling protein: Insights into Parkinson’s disease pathogenesis. Exp. Neurol. 2014, 261, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Minakaki, G.; Krainc, D.; Burbulla, L.F. The Convergence of α-Synuclein, Mitochondrial, and Lysosomal Pathways in Vulnerability of Midbrain Dopaminergic Neurons in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 1465. [Google Scholar] [CrossRef]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from structure to dysfunction: Research progresses, but the themes remain the same. Mol. Neurodegener. 2019, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and α-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.; Imai, Y.; Sokol, N.; Lu, B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Han, K.A.; Shin, W.H.; Jung, S.; Seol, W.; Seo, H.; Ko, C.; Chung, K.C. Leucine-rich repeat kinase 2 exacerbates neuronal cytotoxicity through phosphorylation of histone deacetylase 3 and histone deacetylation. Hum. Mol. Genet. 2017, 26, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-L.; Wu, R.-M. LRRK 2 gene mutations in the pathophysiology of the ROCO domain and therapeutic targets for Parkinson’s disease: A review. J. Biomed. Sci. 2018, 25, 52. [Google Scholar] [CrossRef] [Green Version]
- Trabzuni, D.; Ryten, M.; Emmett, W.; Ramasamy, A.; Lackner, K.J.; Zeller, T.; Walker, R.; Smith, C.; Lewis, P.; Mamais, A.; et al. Fine-Mapping, Gene Expression and Splicing Analysis of the Disease Associated LRRK2 Locus. PLoS ONE 2013, 8, e70724. [Google Scholar] [CrossRef]
- Galter, D.; Westerlund, M.; Carmine, A.; Lindqvist, E.; Sydow, O.; Olson, L. LRRK2 expression linked to dopamine-innervated areas. Ann. Neurol. 2006, 59, 714–719. [Google Scholar] [CrossRef]
- Sharma, S.; Bandopadhyay, R.; Lashley, T.; Renton, A.E.M.; Kingsbury, A.E.; Kumaran, R.; Kallis, C.; Vilariño-Güell, C.; O’Sullivan, S.S.; Lees, A.J.; et al. LRRK2 expression in idiopathic and G2019S positive Parkinson’s disease subjects: A morphological and quantitative study: LRRK2 expression in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2011, 37, 777–790. [Google Scholar] [CrossRef]
- Ferrari, E.; Gandellini, P. Unveiling the ups and downs of miR-205 in physiology and cancer: Transcriptional and post-transcriptional mechanisms. Cell Death Dis. 2020, 11, 980. [Google Scholar] [CrossRef]
- Camps, C.; Saini, H.K.; Mole, D.R.; Choudhry, H.; Reczko, M.; Guerra-Assunção, J.A.; Tian, Y.-M.; Buffa, F.M.; Harris, A.L.; Hatzigeorgiou, A.G.; et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol. Cancer 2014, 13, 28. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, M.; Zhao, K.; He, M.; Ge, W.; Sun, Y.; Wang, Y.; Sun, H.; Yihua, W. Upregulation of MiR-205 under hypoxia promotes epithelial–mesenchymal transition by targeting ASPP2. Cell Death Dis. 2016, 7, e2517. [Google Scholar] [CrossRef]
- Hoover, A.R.; Dozmorov, I.; MacLeod, J.; Du, Q.; De la Morena, M.T.; Forbess, J.; Guleserian, K.; Cleaver, O.B.; Van Oers, N.S.C. MicroRNA-205 Maintains T Cell Development following Stress by Regulating Forkhead Box N1 and Selected Chemokines. J. Biol. Chem. 2016, 291, 23237–23247. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Song, W. Regulation of LRRK2 promoter activity and gene expression by Sp1. Mol. Brain 2016, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, X.; Guo, Y.; Rong, H.; Liu, T. The long noncoding RNA HOTAIR promotes Parkinson’s disease by upregulating LRRK2 expression. Oncotarget 2017, 8, 24449–24456. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; James, W.; Cowley, S. LRRK2 in peripheral and central nervous system innate immunity: Its link to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef]
- Badanjak, K.; Mulica, P.; Smajic, S.; Delcambre, S.; Tranchevent, L.-C.; Diederich, N.; Rauen, T.; Schwamborn, J.C.; Glaab, E.; Cowley, S.A.; et al. iPSC-Derived Microglia as a Model to Study Inflammation in Idiopathic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 740758. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Kamienieva, I.; Duszyński, J.; Szczepanowska, J. Multitasking guardian of mitochondrial quality: Parkin function and Parkinson’s disease. Transl. Neurodegener. 2021, 10, 5. [Google Scholar] [CrossRef]
- Murillo-González, F.E.; García-Aguilar, R.; Vega, L.; Elizondo, G. Regulation of Parkin expression as the key balance between neural survival and cancer cell death. Biochem. Pharmacol. 2021, 190, 114650. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Lesage, S.; Magali, P.; Lohmann, E.; Lacomblez, L.; Teive, H.; Janin, S.; Cousin, P.-Y.; Dürr, A.; Brice, A. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum. Mutat. 2007, 28, 27–32. [Google Scholar] [CrossRef]
- Pankratz, N.; Kissell, D.K.; Pauciulo, M.W.; Halter, C.A.; Rudolph, A.; Pfeiffer, R.F.; Marder, K.S.; Foroud, T.; Nichols, W.C.; For the Parkinson Study Group-PROGENI Investigators. Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 2009, 73, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Itier, J.-M.; Ibáñez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L.; et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; et al. Parkin Protects against the Toxicity Associated with Mutant α-Synuclein: Proteasome Dysfunction Selectively Affects Catecholaminergic Neurons. Neuron 2002, 36, 1007–1019. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Traini, R.; Killinger, B.; Schneider, B.; Moszczynska, A. Overexpression of parkin in the rat nigrostriatal dopamine system protects against methamphetamine neurotoxicity. Exp. Neurol. 2013, 247, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Staropoli, J.F.; McDermott, C.; Martinat, C.; Schulman, B.; Demireva, E.; Abeliovich, A. Parkin Is a Component of an SCF-like Ubiquitin Ligase Complex and Protects Postmitotic Neurons from Kainate Excitotoxicity. Neuron 2003, 37, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S -Nitrosylation of Parkin Regulates Ubiquitination and Compromises Parkin’s Protective Function. Science 2004, 304, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Ko, H.S.; Thomas, B.; Tsang, F.; Chew, K.C.; Tay, S.-P.; Ho, M.W.; Lim, T.-M.; Soong, T.-W.; Pletnikova, O.; et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet. 2005, 14, 3885–3897. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Iemmolo, R.; D’Agata, V.; Scuderi, S.; Drago, F.; Zappia, M.; Cavallaro, S. Increasing the Coding Potential of Genomes Through Alternative Splicing: The Case of PARK2 Gene. Curr. Genom. 2014, 15, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuderi, S.; La Cognata, V.; Drago, F.; Cavallaro, S.; D’Agata, V. Alternative Splicing Generates Different Parkin Protein Isoforms: Evidences in Human, Rat, and Mouse Brain. BioMed Res. Int. 2014, 2014, e690796. [Google Scholar] [CrossRef] [PubMed]
- Mockenhaupt, S.; Makeyev, E.V. Non-coding functions of alternative pre-mRNA splicing in development. Semin. Cell Dev. Biol. 2015, 47–48, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Pisignano, G.; Ladomery, M. Epigenetic Regulation of Alternative Splicing: How LncRNAs Tailor the Message. Non-Coding RNA 2021, 7, 21. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Maugeri, G.; Reitano, R.; Cavallaro, S.; D’Agata, V. Proteomic Analysis of Parkin Isoforms Expression in Different Rat Brain Areas. J. Protein Chem. 2016, 35, 354–362. [Google Scholar] [CrossRef]
- Kasap, M.; Akpinar, G.; Sazci, A.; Idrisoglu, H.A.; Vahaboğlu, H. Evidence for the presence of full-length PARK2 mRNA and Parkin protein in human blood. Neurosci. Lett. 2009, 460, 196–200. [Google Scholar] [CrossRef]
- Shires, S.E.; Quiles, J.M.; Najor, R.H.; Leon, L.J.; Cortez, M.Q.; Lampert, M.A.; Mark, A.; Gustafsson, B. Nuclear Parkin Activates the ERRα Transcriptional Program and Drives Widespread Changes in Gene Expression Following Hypoxia. Sci. Rep. 2020, 10, 8499. [Google Scholar] [CrossRef]
- Brudek, T.; Winge, K.; Rasmussen, N.B.; Bahl, J.M.C.; Tanassi, J.T.; Agander, T.K.; Hyde, T.M.; Pakkenberg, B. Altered α-synuclein, parkin, and synphilin isoform levels in multiple system atrophy brains. J. Neurochem. 2016, 136, 172–185. [Google Scholar] [CrossRef]
- Cheng, M.; Liu, L.; Lao, Y.; Liao, W.; Liao, M.; Luo, X.; Wu, J.; Xie, W.; Zhang, Y.; Xu, N. MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget 2016, 7, 42274–42287. [Google Scholar] [CrossRef] [Green Version]
- Di Rita, A.; Maiorino, T.; Bruqi, K.; Volpicelli, F.; Bellenchi, G.C.; Strappazzon, F. miR-218 Inhibits Mitochondrial Clearance by Targeting PRKN E3 Ubiquitin Ligase. Int. J. Mol. Sci. 2020, 21, 355. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhao, Y.; Li, Z.; Zhu, M.; Wang, Z.; Li, Y.; Xu, T.; Feng, D.; Zhang, S.; Tang, F.; et al. miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol. Res. 2020, 160, 105197. [Google Scholar] [CrossRef]
- Rango, M.; Dossi, G.; Squarcina, L.; Bonifati, C. Brain Mitochondrial Impairment in Early-Onset Parkinson’s Disease With or Without PINK1 Mutation. Mov. Disord. 2020, 35, 504–507. [Google Scholar] [CrossRef]
- Fedorowicz, M.A.; De Vries-Schneider, R.L.A.; Rüb, C.; Becker, D.; Huang, Y.; Zhou, C.; Wolken, D.M.A.; Voos, W.; Liu, Y.; Przedborski, S. Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2014, 15, 86–93. [Google Scholar] [CrossRef]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.-X.; Kim, C.; Nelson, P.T.; Springer, W.; Kim, J. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2013, 2013, e683920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Sullards, M.C.; Olzmann, J.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.-S.; Li, L. Oxidative Damage of DJ-1 Is Linked to Sporadic Parkinson and Alzheimer Diseases. J. Biol. Chem. 2006, 281, 10816–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Lin, S.; Pan, H.; Shen, R.; Wang, M.; Liu, Z.; Sun, S.; Tan, Y.; Wang, Y.; Chen, S.; et al. Lack of Association Between DJ-1 Gene Promoter Polymorphism and the Risk of Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, T.; Liu, W.; Chen, Y. miR-494-3p modulates the progression of in vitro and in vivo Parkinson’s disease models by targeting SIRT3. Neurosci. Lett. 2018, 675, 23–30. [Google Scholar] [CrossRef]
- Siebert, M.; Westbroek, W.; Chen, Y.-C.; Moaven, N.; Li, Y.; Velayati, A.; Saraiva-Pereira, M.L.; E Martin, S.; Sidransky, E. Identification of miRNAs that modulate glucocerebrosidase activity in Gaucher disease cells. RNA Biol. 2015, 11, 1291–1300. [Google Scholar] [CrossRef] [Green Version]
- Straniero, L.; Rimoldi, V.; Samarani, M.; Goldwurm, S.; Di Fonzo, A.; Krüger, R.; Deleidi, M.; Aureli, M.; Soldà, G.; Duga, S.; et al. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci. Rep. 2017, 7, 12702. [Google Scholar] [CrossRef] [Green Version]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, S.M.; Qin, H.; Hendrickson, R.C.; Muwanguzi, J.E.; Lefkowitz, E.J.; Kennedy, R.E.; Yan, Z.; Yacoubian, T.A.; Benveniste, E.N.; West, A.B.; et al. Sex-based differences in the activation of peripheral blood monocytes in early Parkinson disease. npj Park. Dis. 2021, 7, 36. [Google Scholar] [CrossRef]
- Xie, N.; Cui, H.; Banerjee, S.; Tan, Z.; Salomao, R.; Fu, M.; Abraham, E.; Thannickal, V.J.; Liu, G. miR-27a Regulates Inflammatory Response of Macrophages by Targeting IL-10. J. Immunol. 2014, 193, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Zeng, J. Inhibition of miR-494-3p alleviates oxidative stress-induced cell senescence and inflammation in the primary epithelial cells of COPD patients. Int. Immunopharmacol. 2021, 92, 107044. [Google Scholar] [CrossRef]
- Mendizabal, I.; Keller, T.E.; Zeng, J.; Yi, S.V. Epigenetics and Evolution. Integr. Comp. Biol. 2014, 54, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Kriks, S.; Shim, J.-W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- De Boni, L.; Wüllner, U. Epigenetic Analysis in Human Neurons: Considerations for Disease Modeling in PD. Front. Neurosci. 2019, 13, 276. [Google Scholar] [CrossRef] [Green Version]
- Rando, T.A.; Chang, H.Y. Aging, Rejuvenation, and Epigenetic Reprogramming: Resetting the Aging Clock. Cell 2012, 148, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Reid, D.; Lau, S.; Kim, Y.; Gage, F.H. Aging in a Dish: iPSC-Derived and Directly Induced Neurons for Studying Brain Aging and Age-Related Neurodegenerative Diseases. Annu. Rev. Genet. 2018, 52, 271–293. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2018, 20, 109–127. [Google Scholar] [CrossRef]
- Jost, D.; Vaillant, C. Epigenomics in 3D: Importance of long-range spreading and specific interactions in epigenomic maintenance. Nucleic Acids Res. 2018, 46, 2252–2264. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Zhang, B. Predicting three-dimensional genome organization with chromatin states. PLoS Comput. Biol. 2019, 15, e1007024. [Google Scholar] [CrossRef] [Green Version]
- Rhie, S.K.; Perez, A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies | Tissues Analyzed | Proteins in Controls vs. sPD | mRNA in Controls vs. sPD | DNA Methylation in Controls vs. sPD | MiRNA Expression in Controls vs. sPD | Reference |
|---|---|---|---|---|---|---|
| SNCA | ||||||
| Grundemann et al., 2008 | Brain: DA neurons of SN | Increase | Increase | - | - | [16] |
| Jowaed et al., 2010/Matsumoto et al., 2010 | Brain | - | - | Hypomethylation | - | [17,18] |
| Pihlstrom et al., 2015/Ai et al., 2014/Tan et al., 2014 | Blood immune cells | - | - | Hypomethylation | - | [19,20,21] |
| Minones-Moyano et al., 2011 | Brain | - | - | - | Decrease miR-34b/c | [22] |
| LRRK2 | ||||||
| Cho et al., 2013 | Brain: frontal cortex/striatum | Increase | No difference | - | Decrease miR-205 | [23] |
| Cook et al., 2017 | Blood | Increase | - | - | - | [24] |
| Tan et al., 2014 | Blood immune cells | - | - | Hypomethylation | - | [21] |
| PRKN | ||||||
| Beyer et al., 2008 | Brain | - | Increase in variant TV3 and TV12 | - | - | [25] |
| Cai et al., 2011 | Blood immune cells | - | - | No difference | - | [26] |
| De Mena et al., 2013 | Brain | - | - | No difference | - | [27] |
| Eryilmaz et al., 2017 | Blood immune cells | - | - | Hypomethylation | - | [28] |
| Ding et al., 2016 | Plasma | - | - | - | Decrease miR-181a | [29] |
| Xing et al., 2020 | Brain | - | - | - | Decrease miR-218 | [30] |
| Serafin et al., 2015 | Plasma | - | - | - | Increase miR-103a-3p | [31] |
| PINK1 | ||||||
| Muqit et al., 2006 | Brain | Increase ∆1-PINK1 | - | - | - | [32] |
| Blackinton et al., 2007 | Brain: SN | - | No difference | - | - | [33] |
| Navarro-Sanchez et al., 2018 | Brain: SN | - | - | No difference | - | [34] |
| Fazeli et al., 2020/Dos Santos et al., 2018 | PBMC/CSF | - | - | - | Decrease miR-27a | [35,36] |
| DJ1 | ||||||
| Kumaran et al., 2009 | Brain | Decrease | Decrease | - | - | [37] |
| Tan et al., 2016 | Blood immune cells | - | - | No difference | - | [38] |
| Chen et al., 2017 | Plasma | - | - | - | Increase miR-4639-5p | [39] |
| GBA | ||||||
| Murphy et al., 2014 | Brain | Decrease | No difference | - | - | [40] |
| Moors et al., 2019 | Brain | - | No difference | - | - | [41] |
| Eryilmaz et al., 2017 | Blood immune cells | - | - | No difference | - | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanore, A.; Lesage, S.; Mariani, L.-L.; Menon, P.J.; Ravassard, P.; Cheval, H.; Corti, O.; Brice, A.; Corvol, J.-C. Does the Expression and Epigenetics of Genes Involved in Monogenic Forms of Parkinson’s Disease Influence Sporadic Forms? Genes 2022, 13, 479. https://doi.org/10.3390/genes13030479
Lanore A, Lesage S, Mariani L-L, Menon PJ, Ravassard P, Cheval H, Corti O, Brice A, Corvol J-C. Does the Expression and Epigenetics of Genes Involved in Monogenic Forms of Parkinson’s Disease Influence Sporadic Forms? Genes. 2022; 13(3):479. https://doi.org/10.3390/genes13030479
Chicago/Turabian StyleLanore, Aymeric, Suzanne Lesage, Louise-Laure Mariani, Poornima Jayadev Menon, Philippe Ravassard, Helene Cheval, Olga Corti, Alexis Brice, and Jean-Christophe Corvol. 2022. "Does the Expression and Epigenetics of Genes Involved in Monogenic Forms of Parkinson’s Disease Influence Sporadic Forms?" Genes 13, no. 3: 479. https://doi.org/10.3390/genes13030479