
Comparing BeadChip and WGS Genotyping: Non-Technical Failed Calling Is Attributable to Additional Variation within the Probe Target Sequence


Abstract
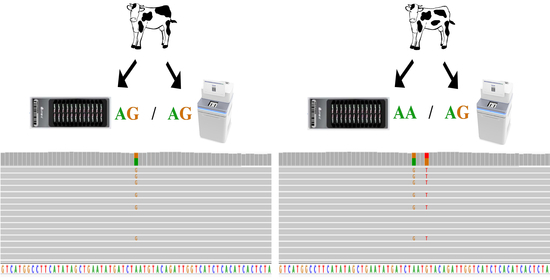
:
1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Sample Preparation for WGS
2.2. WGS Analysis for the VCF Method
2.3. WGS Analysis for GAP5 Method
2.4. Statistics
3. Results
3.1. Selection of Polymorphic Sites and Genomic Coordinates
3.2. WGS and Variant Calling from Next-Generation Sequencing (NGS) Data
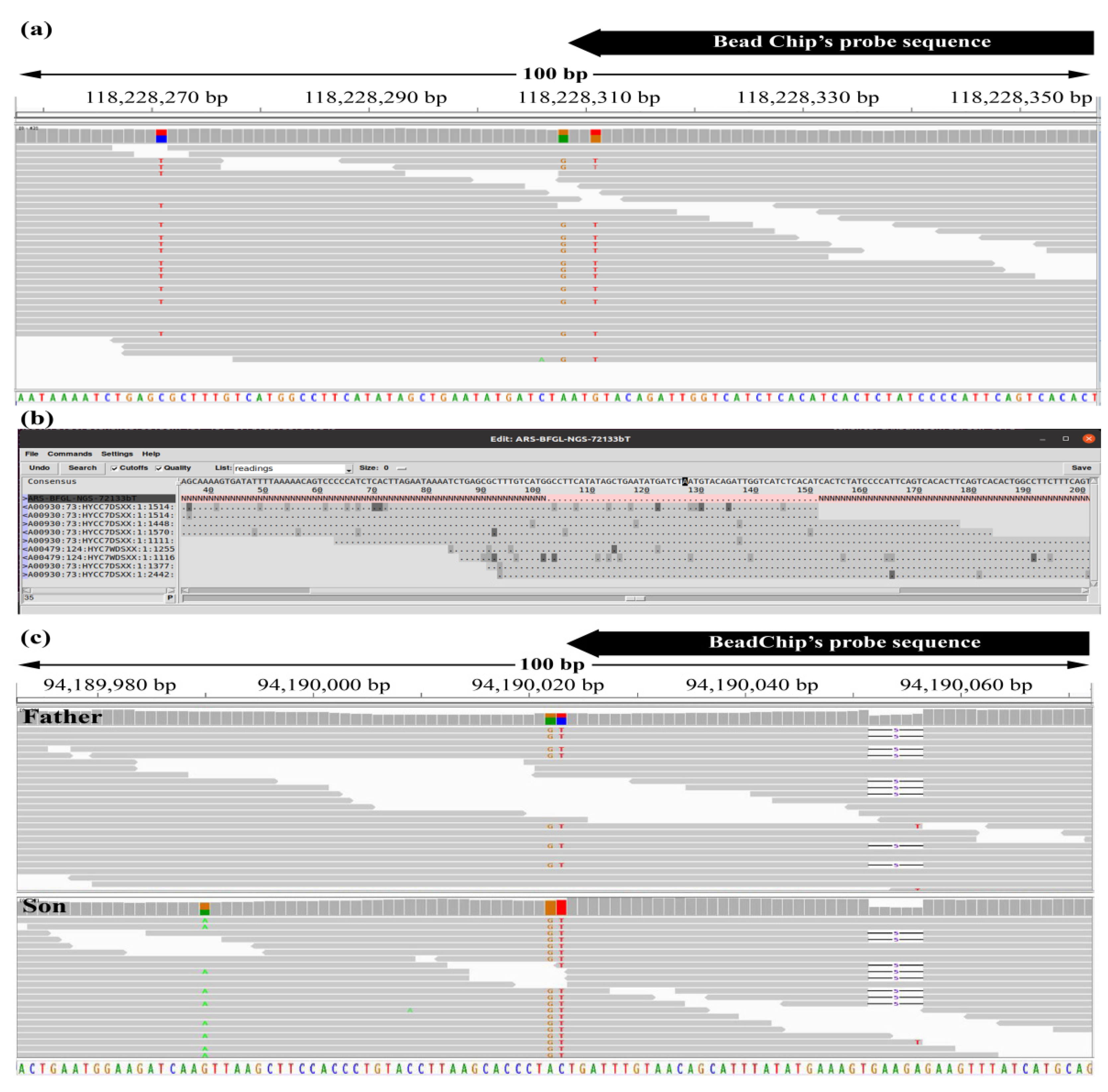
3.3. Case Analyses of Beadchip and WGS Genotype Mismatches
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parnell, P. Introduction to DNA technology-what will it offer the beef industry? In Proceedings of the Beef Products Program: Technology-Our Future, Paterson, NSW, Australia, 13 May 2003; pp. 57–160. [Google Scholar]
- Grisart, B.; Coppieters, W.; Farnir, F.; Karim, L.; Ford, C.; Berzi, P.; Cambisano, N.; Mni, M.; Reid, S.; Simon, P.; et al. Positional candidate cloning of a QTL in dairy cattle: Identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002, 12, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Zinder, M.; Seroussi, E.; Larkin, D.M.; Loor, J.J.; Everts-van der Wind, A.; Lee, J.H.; Drackley, J.K.; Band, M.R.; Hernandez, A.G.; Shani, M.; et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005, 15, 936–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.I.; Ezra, E.; Ron, M. Invited review: A perspective on the future of genomic selection in dairy cattle. J. Dairy Sci. 2017, 100, 8633–8644. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.W.; Van Tassell, C.P.; VanRaden, P.M.; Wiggans, G.R. Four ways genomic selection will change dairy cattle genetic improvement in the near future. Progress. Dairym. 2008, 22, 14–17. [Google Scholar]
- VanRaden, P.M.; O’Connell, J.R.; Wiggans, G.R.; Weigel, K.A. Genomic evaluations with many more genotypes. Genet. Sel. Evol. 2011, 43, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, K.L.; Steemers, F.J.; Lee, G.; Mendoza, L.G.; Chee, M.S. A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet. 2005, 37, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.; Freudenberg, J.; Thompson, S.; Aronow, B.; Pavlidis, P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res. 2005, 33, 5914–5923. [Google Scholar] [CrossRef] [Green Version]
- Van Tassell, C.P.; Smith, T.P.; Matukumalli, L.K.; Taylor, J.F.; Schnabel, R.D.; Lawley, C.T.; Haudenschild, C.D.; Moore, S.S.; Warren, W.C.; Sonstegard, T.S. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods 2008, 5, 247–252. [Google Scholar] [CrossRef]
- Seroussi, E.; Glick, G.; Shirak, A.; Yakobson, E.; Weller, J.I.; Ezra, E.; Zeron, Y. Analysis of copy loss and gain variations in Holstein cattle autosomes using BeadChip SNPs. BMC Genom. 2010, 11, 673. [Google Scholar] [CrossRef] [Green Version]
- Wiggans, G.R.; Cooper, T.A.; VanRaden, P.M.; Van Tassell, C.P.; Bickhart, D.M.; Sonstegard, T.S. Increasing the number of single nucleotide polymorphisms used in genomic evaluation of dairy cattle. J. Dairy Sci. 2016, 99, 4504–4511. [Google Scholar] [CrossRef]
- Danilov, K.A.; Nikogosov, D.A.; Musienko, S.V.; Baranova, A.V. A comparison of BeadChip and WGS genotyping outputs using partial validation by sanger sequencing. BMC Genom. 2020, 21, 528. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Zimin, A.; Dreischer, C.; Schultheiss, S.; Hall, R.; Schroeder, S.G.; et al. Modernizing the bovine reference genome assembly. In Proceedings of the 11th World Congress on Genetics Applied to Livestock Production, Hamilton, New Zealand, 7 February 2018. [Google Scholar]
- Arafat, M.; Kleiman, S.E.; AbuMadighem, A.; Zeadna, A.; Levitas, E.; Vardi, I.H.; Barda, S.; Lehavi, O.; Hauser, R.; Lunenfeld, E.; et al. Pathogenic variations in Germ Cell Nuclear Acidic Peptidase (GCNA) are associated with human male infertility. Eur. J. Hum. Genet. 2021, 29, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, R.D. ARS-UCD1.2 Cow Genome Assembly: Mapping of All Existing Variants. Available online: https://www.animalgenome.org/repository/cattle/UMC_bovine_coordinates/UMC_marker_names_180910.zip (accessed on 10 February 2022).
- Pirooznia, M.; Kramer, M.; Parla, J.; Goes, F.S.; Potash, J.B.; McCombie, W.R.; Zandi, P.P. Validation and assessment of variant calling pipelines for next-generation sequencing. Hum. Genom. 2014, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Dapprich, J.; Ferriola, D.; Mackiewicz, K.; Clark, P.M.; Rappaport, E.; D’Arcy, M.; Sasson, A.; Gai, X.; Schug, J.; Kaestner, K.H.; et al. The next generation of target capture technologies-large DNA fragment enrichment and sequencing determines regional genomic variation of high complexity. BMC Genom. 2016, 17, 486. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.S.; Smith, J.D.; Stanaway, I.B.; Rieder, M.J.; Nickerson, D.A. Direct detection of null alleles in SNP genotyping data. Hum. Mol. Genet. 2006, 15, 1931–1937. [Google Scholar] [CrossRef] [Green Version]
- Franke, L.; de Kovel, C.G.; Aulchenko, Y.S.; Trynka, G.; Zhernakova, A.; Hunt, K.A.; Blauw, H.M.; van den Berg, L.H.; Ophoff, R.; Deloukas, P.; et al. Detection, imputation, and association analysis of small deletions and null alleles on oligonucleotide arrays. Am. J. Hum. Genet. 2008, 82, 1316–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abo-Ismail, M.K.; Brito, L.F.; Miller, S.P.; Sargolzaei, M.; Grossi, D.A.; Moore, S.S.; Plastow, G.; Stothard, P.; Nayeri, S.; Schenkel, F.S. Genome-wide association studies and genomic prediction of breeding values for calving performance and body conformation traits in Holstein cattle. Genet. Sel. Evol. 2017, 49, 82. [Google Scholar] [CrossRef] [Green Version]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Park, K.D.; Ka, S.; Kim, H.; Cho, K.H. Heritability estimated using 50K SNPs Indicates missing heritability problem in Holstein breeding. Genom. Inform. 2015, 13, 146–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.; Lee, S.H.; Robinson, M.R.; Perry, J.R.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V.; et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015, 47, 1114–1120. [Google Scholar] [CrossRef]
- Maroti, Z.; Boldogkoi, Z.; Tombacz, D.; Snyder, M.; Kalmar, T. Evaluation of whole exome sequencing as an alternative to BeadChip and whole genome sequencing in human population genetic analysis. BMC Genom. 2018, 19, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, R.; Berg, I.V.D.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. USA 2019, 116, 19398–19408. [Google Scholar] [CrossRef] [Green Version]
- Xiang, R.; MacLeod, I.M.; Daetwyler, H.D.; de Jong, G.; O’Connor, E.; Schrooten, C.; Chamberlain, A.J.; Goddard, M.E. Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations. Nat. Commun. 2021, 12, 860. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Sire | NGS 1 | # of Spots 2 | Match GAP5 3 | Match VCF 3 | Miss GAP5 4 | Miss VCF 4 |
|---|---|---|---|---|---|---|
| 3376 | NS | 401,967,974 | 34,493 | 35,030 | 3 | 25 |
| 3651 | NS | 361,519,877 | 34,452 | 35,081 | 3 | 13 |
| 3756 | NS | 449,885,842 | 34,507 | 35,033 | 6 | 18 |
| 3811 | NS | 399,980,764 | 34,543 | 35,061 | 0 | 17 |
| 7165 | NS | 382,156,099 | 34,397 | 35,055 | 5 | 21 |
| 7592 | NS | 458,256,777 | 33,397 | 33,796 | 0 | 9 |
| 7733 | NS | 439,943,852 | 42,542 | 38,845 | 16 | 81 |
| Mean ± SE 5 | 413,387,312 ± 13,824,080 | 35,476 ± 1188 | 35,415 ± 599 | 4.7 ± 2.1 | 26.3 ± 9.3 | |
| 7396 | HS | 310,285,759 | 44,238 | 40,753 | 15 | 54 |
| 7400 | HS | 319,575,305 | 43,364 | 41,313 | 5 | 67 |
| 7424 | HS | 337,946,649 | 44,879 | 41,136 | 5 | 57 |
| 7510 | HS | 300,382,216 | 44,890 | 41,376 | 2 | 62 |
| 7559 | HS | 273,572,927 | 44,626 | 41,221 | 6 | 62 |
| 7679 | HS | 285,115,013 | 43,762 | 40,470 | 8 | 62 |
| 7733 | HS | 302,259,900 | 42,224 | 38,849 | 16 | 78 |
| 7738 | HS | 296,384,014 | 44,778 | 41,334 | 6 | 76 |
| 7851 | HS | 327,126,812 | 44,945 | 41,320 | 5 | 56 |
| 7936 | HS | 317,888,134 | 44,721 | 41,154 | 7 | 56 |
| 9078 | HS | 304,472,448 | 44,890 | 41,355 | 10 | 64 |
| Mean ± SE 5 | 306,819,016 ± 5,585,356 | 44,302 ± 260 | 40,934 ± 225 | 7.7 ± 1.3 | 63.1 ± 2.4 |
| Total | Polymorphic | Variation Upstream | Variation Downstream | Any Extra Variation | Not SNP 2 | Potential Problem | |
|---|---|---|---|---|---|---|---|
| # of SNPs | 50,392 | 42,848 | 10,381 | 10,237 | 17,197 | 393 | 17,265 |
| % | 100 | 85 | 24.2 | 23.9 | 40.1 | 0.9 | 40.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gershoni, M.; Shirak, A.; Raz, R.; Seroussi, E. Comparing BeadChip and WGS Genotyping: Non-Technical Failed Calling Is Attributable to Additional Variation within the Probe Target Sequence. Genes 2022, 13, 485. https://doi.org/10.3390/genes13030485
Gershoni M, Shirak A, Raz R, Seroussi E. Comparing BeadChip and WGS Genotyping: Non-Technical Failed Calling Is Attributable to Additional Variation within the Probe Target Sequence. Genes. 2022; 13(3):485. https://doi.org/10.3390/genes13030485
Chicago/Turabian StyleGershoni, Moran, Andrey Shirak, Rotem Raz, and Eyal Seroussi. 2022. "Comparing BeadChip and WGS Genotyping: Non-Technical Failed Calling Is Attributable to Additional Variation within the Probe Target Sequence" Genes 13, no. 3: 485. https://doi.org/10.3390/genes13030485