Highlights
What are the main findings?
- Review of KMT2A germline mutations associated with rare genetic disorders that affect the epigenetic apparatus and neurodevelopment (i.e., chromatinopathies).
- Review of KMT2A somatic mutations in all the different types of tumors with their relative frequency.
- Review of in vivo models with KMT2A alterations dissecting their effect on the animal phenotype.
- Review of therapeutic approaches targeting the regulators of lysine methylation.
What is the implication of the main finding?
- KMT2A germline mutations characterize patients with Wiedemann–Steiner syndrome but were also reported in patients with initial clinical diagnoses of other chromatinopathies with overlapping phenotypes.
- KMT2A somatic mutations are present in several tumors; interestingly, the most recurrent somatic mutation types differ from the germline ones.
- KMT2A-depleted animal models recapitulate phenotypes described for patients with both germline and somatic mutations, exhibiting hematological and neurodevelopmental defects.
- Acting on the regulators of lysine methylation could represent an epigenetic intervention that is either pharmaceutical or nutritional for both cancer treatment and the amelioration of defective cognitive/behavioral traits in rare diseases.
Abstract
KMT2A (Lysine methyltransferase 2A) is a member of the epigenetic machinery, encoding a lysine methyltransferase responsible for the transcriptional activation through lysine 4 of histone 3 (H3K4) methylation. KMT2A has a crucial role in gene expression, thus it is associated to pathological conditions when found mutated. KMT2A germinal mutations are associated to Wiedemann–Steiner syndrome and also in patients with initial clinical diagnosis of several other chromatinopathies (i.e., Coffin–Siris syndromes, Kabuki syndrome, Cornelia De Lange syndrome, Rubinstein–Taybi syndrome), sharing an overlapping phenotype. On the other hand, KMT2A somatic mutations have been reported in several tumors, mainly blood malignancies. Due to its evolutionary conservation, the role of KMT2A in embryonic development, hematopoiesis and neurodevelopment has been explored in different animal models, and in recent decades, epigenetic treatments for disorders linked to KMT2A dysfunction have been extensively investigated. To note, pharmaceutical compounds acting on tumors characterized by KMT2A mutations have been formulated, and even nutritional interventions for chromatinopathies have become the object of study due to the role of microbiota in epigenetic regulation.
1. Introduction
KMT2A (Lysine methyltransferase 2A), also known as MLL1, is a protein coding gene mapping to human chromosome 11 (11q23.3), made up of 90,343 bases (GRCh38/hg38) and 37 exons belonging to KMTs (Lysine methyltransferases) family.
KMTs catalyze the transfer of methyl groups from S-adenosylmethionine to the lysine residues on histone tails, particularly the histone H3 tail. Unlike other epigenetic enzymes such as acetyltransferases (HATs), KMTs are more specific and usually modify one or two lysines on a single histone [1]. Lysines can be monomethylated, bimethylated or trimethylated without changing the electric charge of the amino acid side chain. The effect on chromatin state, i.e., whether it activates transcription or represses it, depends on the methylation states and their positions (Figure 1) [2,3,4,5,6,7,8,9,10,11,12,13,14,15]. KMTs are so called writers, enzymes that catalyze the addition of chemical groups to histone tails or to DNA; these modifications are not permanent but can be removed by erasers to reverse the influence on gene expression. Readers possess specialized domains able to recognize and interpret different chemical modifications. Writers, erasers and readers form the epigenetic machinery, and mutations in genes coding for this apparatus lead to ann altered chromatin conformation and an incorrect gene expression, resulting in a series of syndromes known as chromatinopathies, Mendelian genetic diseases, most of them with a dominant character [16,17,18]. Pathogenic mutations in KMTs and KDMs (Lysine demethylases) lead to haploinsufficiency in numerous developmental syndromes (Figure 2) (Table 1) [10,19].
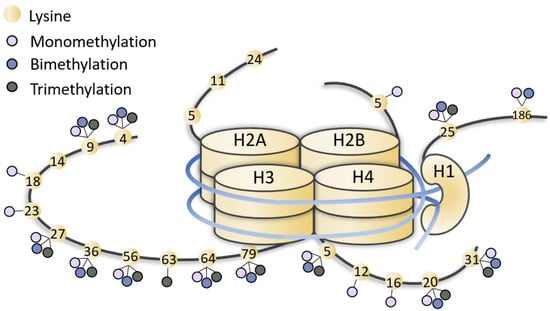
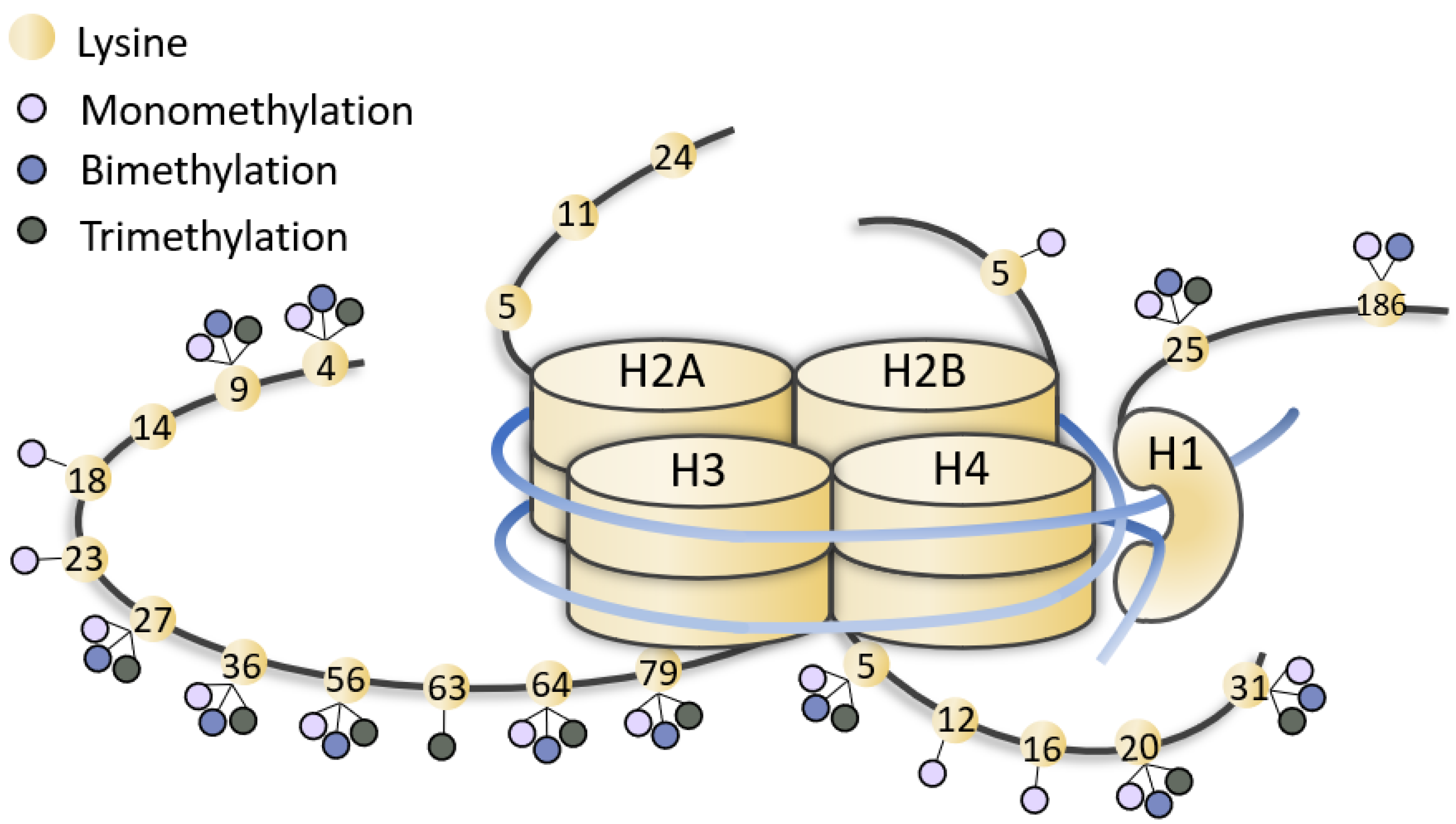
Figure 1.
Representation of methylated lysines of histone tails. Lysines (yellow dots) of histone (H1, H2, H3, H4) tails can be mono-, bi-, tri-methylated (little lilac, blue and grey dots). The figure is not drawn to scale.
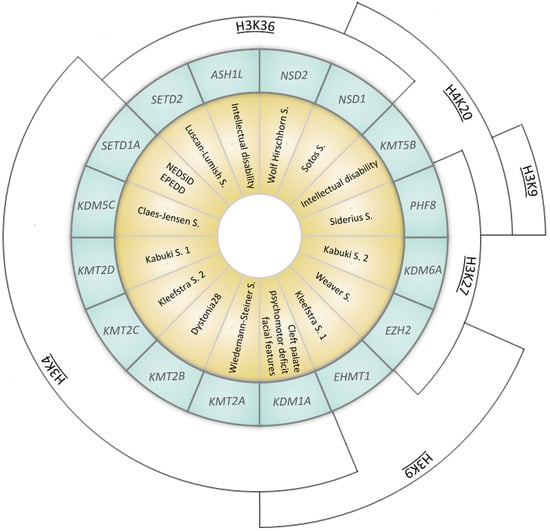
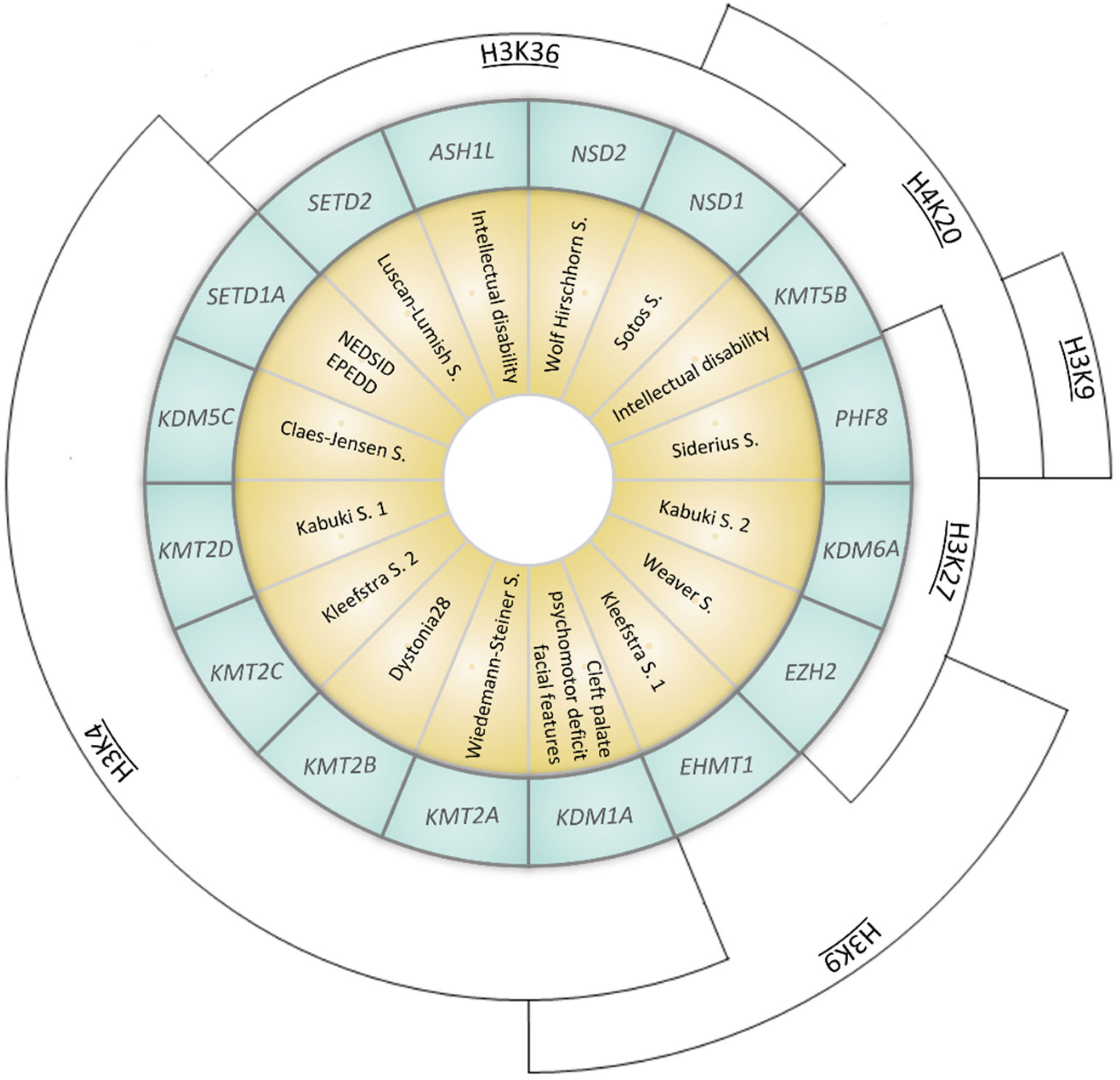
Figure 2.
Representation of syndromes caused by mutations in genes coding for KMTs (Lysine methyltransferases) or KDMs (Lysisne demethylases). Syndromes (yellow inner ring) and the corresponding causative gene (coding for KMTs or KDMs, listed in the middle blue ring) are represented. The outer arcs indicate the site of epigenetic modification (NEDSID: Neurodevelopmental disorder with speech impairment and dysmorphic facies; EPEDD: Epilepsy, early-onset, with or without developmental delay).

Table 1.
Details of genes and syndromes represented in Figure 2.
Many species have a KMT2A ortholog, including fishes, birds, amphibians, and mammals; thus, its evolutionary conservation allowed a comprehensive study of KMT2A molecular functions through in vivo experiments on animal models (Drosophila melanogaster, Danio rerio, Mus musculus). KMT2A expression is mainly nuclear and ubiquitously present in 27 tissues, especially in ovary, lymph node, endometrium, thyroid and brain tissue [20]. KMT2A encodes a lysine methyltransferase (KMT) formed of 3969 amino acids, a transcriptional co-activator which plays a crucial role in hematopoiesis, in regulating gene expression at early developmental stages, and in the control of circadian gene expression. KMT2A is processed by the endopeptidase Taspase 1 in two fragments (MLL-C and MLL-N) which heterodimerize and regulate the transcription of specific genes, including HOX genes [21]. KMT2A protein has 18 domains, including the CXXC-type zinc finger, the extended PHD domain and the bromodomain. The SET domain has the methyltransferase activity (mono-, di-, tri-methylation) on lysine 4 of histone 3 (H3K4 me1/2/3), a post-transcriptional modification (PTM) responsible of epigenetic transcriptional activation and which efficiency can be increased when the protein is associated with another component of the MLL1/MLL complex (Figure 3) [22].
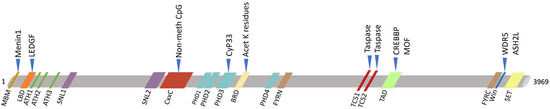
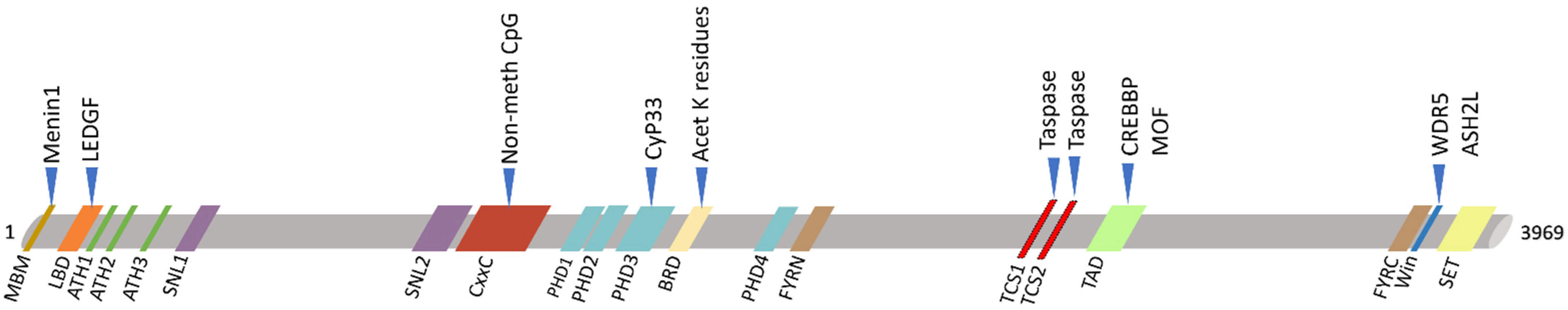
Figure 3.
Schematic view of KMT2A protein domains (below) and its main interactors (upper). KMT2A domains: MBM, high-affinity Menin-binding motif, residues 6–10; LBD, LEDGF-binding domain, residues 109–153; ATH1-2-3, AT-Hook1/2/3, residue 169–180, residues 217–227, residue 301–309; SNL1-2, nuclear-localization signal 1/2, residues 400–443, residues 1008–1106; CxxC, including: pre-CxxC region, residues 1149–1154, CxxC domain, residues 1147–1242, post-CxxC residues 1298–1337; PHD1-2-3-4, plant homology domain 1/2/3/4, residues 1431–1482, residues 1479–1533, residues 1566–1627, residues 1931–1978; BRD, bromodomain, residues 1703–1748; FYRN, FY-rich N-terminal domain, residues 2018–2074; TAD, transactivator domain, residues 2829–2883; FYRC, FY-rich C-terminal domain, residues 3666–3747; Win, WDR5 interaction motif, residues 3762–3773; SET, Su(Var)3-9 enhancer-of-zeste trithorax domain, residues 3829–2945. KMT2A has two sites for cutting by Taspase1: TCS1-2, taspase1 cleavage site 1/2, residue 2666–2670 and residues 2718–2722.
As other members of KMTs family, KMT2A regulates gene transcription through chromatin opening or closure and its activity is antagonized by the lysine demethylases (KDMs) family.
2. KMT2A Germline Mutations
2.1. Wiedemann–Steiner Syndrome
KMT2A germinal variants are associated to the Wiedemann–Steiner syndrome (WDSTS, OMIM #605130), a rare autosomal dominant disorder characterized by different features, mainly intellectual disability (ID), developmental delay (DD), pre- and post-natal growth deficiency, hypertrichosis, short stature, hypotonia, distinctive facial features (thick eyebrows, long eyelashes, narrow palpebral fissures, broad nasal tip, down slanting palpebral fissures), skeletal abnormalities (clinodactyly, brachydactyly, accelerated skeletal maturation), feeding problems and behavioral difficulties (Figure 4A) (Table 2) [23,24,25]. KMT2A variants are distributed throughout the gene, with a pathogenic mutation hotspot in exon 27, and most of them lead to KMT2A loss of function. WDSTS patients usually present de novo private mutations, and the diagnosis is based on clinical evaluation of signs and symptoms then confirmed by molecular analysis. Unfortunately, a specific treatment is not available, thus possible interventions aim at reducing the severity of symptoms.

Figure 4.
Comparison of typical facial features of (A) Wiedemann–Steiner syndrome [26]; (B) Coffin–Siris syndrome; (C) Kabuki syndrome; (D) Cornelia De Lange syndrome [27]; (E) Rubinstein–Taybi syndrome [28].
Table 2.
Clinical signs reported in patients with a KMT2A mutation and an initial clinical diagnosis of chromatinopathy. Presence of all features is compared with the one in WDSTS.
2.2. Other Chromatinopathies
Mutations in KMT2A have been also found in patients with a clinical presentation suggestive of other chromatinopathies but negative for alterations in the related known-causative genes. Their clinical presentation shares with WDSTS some phenotypic features and it is caused by alterations of genes involved in the regulation and maintenance of chromatin state as KMT2A. Indeed, these syndromes are caused by mutations in genes of the epigenetic machinery and therefore are known as chromatinopathies [16,18].
In 2015, a whole exome sequencing (WES) analysis of a cohort of 46 individuals with an initial diagnosis of Coffin–Siris syndromes (CSS1, OMIM #135900; CSS2, OMIM #614607; CSS3, OMIM #614608; CSS4, OMIM #614609; CSS5, OMIM #616938; CSS6, OMIM #617808; CSS7, OMIM #618027; CSS8, OMIM #618362; CSS9, OMIM #615866; CSS10, OMIM #618506; CSS11, OMIM #618779; CSS12, OMIM #619325) or Nicolaides–Baraitser syndromes (NCBRS, OMIM #601358) revealed a heterozygous de novo missense mutation in the KMT2A gene in a boy clinically diagnosed with CSS1 [29]. Coffin–Siris syndrome is a rare multisystemic congenital syndrome characterized by developmental or cognitive delay (from mild to severe), congenital anomalies involving different systems such as the genitourinary, cardiac or gastrointestinal (GI)systems or the central nervous system (CNS), distinctive facial features and musculoskeletal anomalies (aplasia or hypoplasia of the distal phalanx or nail of the fifth and additional digits) (Figure 4B) [30]. CSS is caused by mutations in genes encoding subunits of the BAF (ATP-dependent BRG1/BRM associated factor) complex, which functions as a chromatin remodeling factor and includes ARID1B (6q25.3, OMIM #614556; associated with CSS1), ARID1A (1p36.1-p35, OMIM #603024; associated with CSS2), SMARCB1 (22q11.23, OMIM #601607; associated with CSS3), SMARCA4 (19p13.3, OMIM #603254; associated with CSS4), SMARCE1 (17q21.2, OMIM #603111; associated with CSS5), ARID2 (12q12, OMIM #609539; associated with CSS6), DFP (11q13.1, OMIM #601671; associated with CSS7), SMARCC2 (12q13.2, OMIM #601734; associated with CSS8), SOX11 (2p15.2; OMIM #600898; associated with CSS9), SOX4 (6p22.3, OMIM #184430; associated with CSS10), SMARCD1 (12q13.12, OMIM #601735; associated with CSS11) and BICRA (19q13.33, OMIM #605690; associated with CSS12). The CSS1 patient with KMT2A mutation showed cardiac anomalies (patent ductus arteriosus and mitral valve prolapse), right retinal atrophy and unilateral cryptorchidism. He exhibited speech delay and peculiar signs such as bilateral fifth finger clinodactyly and dysmorphisms, including upslanted palpebral fissures, long eyelashes, a bulbous nasal tip, long philtrum and a full lower vermillion. In addition, he suffered from recurrent pulmonary infection [29] (Table 2).
Thanks to targeted sequencing and genome-wide DNA methylation analyses, in 2017, Sobreira and colleagues, investigating a cohort of 27 patients with a clinical diagnosis of Kabuki syndrome (KS1, OMIM #147920; KS2, OMIM #300867), found two patients positive for mutations in KMT2A (a de novo heterozygous missense mutation in pt#KS8 and a donor splice site mutation in pt#KS29) [31]. Kabuki syndrome is a congenital disease with a broad and variable spectrum, characterized by mild-to-moderate cognitive disability, post-natal growth deficit, characteristic facial features (long eyelid cracks with slight ectropion of lateral third of the lower eyelid), skeletal abnormalities and immunodeficiency (Figure 4C) [32]. In about 60% of KS cases, the syndrome is caused by mutations in KMT2D (12q13.12, OMIM #602113; associated with KSS1), also known as MLL2, while in a few cases the causative mutation is carried by the KDM6A gene (Xp11.3, OMIM #300128; associated with KSS2). KMT2D is a methyltransferase that plays crucial roles in development, differentiation, metabolism, and tumor suppression [33]. Both patients analysed by Sobreira and colleagues presented hypotonia, persistent fetal fingerpads, eversion of the lower lateral lid and long palpebral fissure; patient #KS8 in addition showed seizures and recurrent infection and brachydactyly, while patient #KS29 presented ID and feeding difficulties (Table 2) [31].
In two different works, patients with Cornelia De Lange syndrome (CdLS1, OMIM #122470; CdLS2, OMIM #300590; CdLS3, OMIM #610759; CdLS4, OMIM #614701; CdLS5, OMIM #300882)-like phenotype were found carriers of pathogenetic variants in KMT2A. Cornelia De Lange syndrome is a rare and clinically variable neurodevelopmental disorder characterized by ID, distinctive facial features, prenatal and postnatal growth retardation, congenital anomalies (malformations of the upper limbs, gastrointestinal malformation/rotation, heart defects and genitourinary malformations), and behavioral problems (Figure 4D) [34]. So far, mutations leading to CdLS have been identified in seven genes: NIPBL (5p13.2, OMIM #608667; associated with CdLS1), SMC1A (Xp11.22, OMIM #300040; associated with CdLS2), SMC3 (10q25.2, OMIM #606062; associated with CdLS3), RAD21 (8q24.11, OMIM #606462; associated with CdLS4), HDAC8 (Xq13.1, OMIM #300269; associated with CdLS5) and the two more recently described BRD4 (19p13.12) and ANKRD11 (16q24.3). All these genes belong to the cohesin complex involved in chromosome segregation, DNA repair and gene regulation [34]. CdLS5 belongs to the chromatinopathies group, as HDAC8 is a histone deacetylase acting on the chromatin structure with a transcriptional repression effect. In 2015, a WES analysis on 32 Turkish individuals revealed the presence of a de novo heterozygous nonsense KMT2A mutation in one female patient [35]. She presented developmental and growth delay, microcephaly, clinodactyly, hirsutism, DD/ID and facial dysmorphisms including long philtrum, thin and arched eyebrows, synophrys, long eyelashes, a thin upper lip, and a high arched palate (Table 2). Two years later, Parenti and colleagues identified a de novo nonsense mutation in KMT2A in a male patient with CdLS clinical diagnosis, through targeted next-generation sequencing (NGS) analysis [36]. He exhibited growth retardation, mild ID and peculiar dysmorphisms (arched eyebrows with synophrys, long eyelashes, ptosis, bulbous nasal tip and thin upper vermillion border of the lip), while minor anomalies included small hands and clinodactyly of the fifth finger (Table 2).
More recently, two studies identified pathogenetic variants in KMT2A in patients with initial diagnosis of Rubinstein–Taybi syndrome (RSTS1, OMIM #180849; RSTS2; OMIM #613684) [26,37]. RSTS is a highly rare autosomal dominant genetic disorder, characterized by typical facial features, skeletal abnormalities (microcephaly, broad thumbs and first toes), ID, speech delay, and postnatal growth retardation (Figure 4E) [28,38,39]. RSTS is mostly caused (70%) by heterozygous pathogenic variants in CREBBP (16p13.3, OMIM # 600140; associated with RSTS1), or in few cases (10%) in EP300 (22q13.2, OMIM #602700; associated with RSTS2) [40,41]. CREBBP and EP300 encode, respectively, for CBP and p300, two lysin acetyltransferases (KATs) involved in the opening of the chromatin, in consequent transcriptional regulation and fundamental biological pathways [42,43,44,45,46]. In 2019, a WES analysis was carried out on patients clinically diagnosed with RSTS, and one male patient (number #103) was found positive for mutation in KMT2A. He presented hypotonia, skeletal anomalies involving hands and feet, and different facial features such as synophrys, arched and thick eyebrows, and downslanting palpebral fissure among others (Table 2) [37]. In 2021, a study individuated heterozygous variants in KMT2A in six patients with an RSTS-like phenotype but negative for RSTS known causative genes, thanks to the NGS approach (multigene panel sequencing and WES) [26]. The most common features displayed by these patients were ID (6/6), long eyelashes (6/6), speech delay (5/6), broad halluces (5/6), columella below the alae nasi (5/6), wide nasal bridge (4/6), ptosis (4/6), downslanting palpebral fissures (4/6), thick eyebrow (4/6), postnatal growth retardation (4/6) and behavioral problems (4/6). In addition, half of patients presented hirsutism and two of them showed hypotonia and feeding problems (Table 2). Importantly, abundant evidence suggests a clinical and molecular overlap for mutations in genes encoding proteins involved in the regulation and maintenance of the chromatin state. Thus, it is possible to hypothesize a thorough molecular evaluation for shared altered pathways in the future, for a correct diagnosis.
3. KMT2A Somatic Mutations
KMT2A somatic mutations are implicated in several tumors. The most common types of alterations involving KMT2A are mutations (3.62%), fusions (0.13%) (with more than 80 different partners identified) [47], losses (0.10%), amplifications (0.07%), and KMT2A-EP300 fusions (0.19%) [48]. Among the mutations, the most frequent observed in patient-derived samples are missense (54.36%), synonymous (13.61%) and nonsense substitutions (7.34%) [49]. On the contrary, in patients with germline KMT2A mutations, the ones more represented are frameshift (41%) and stop mutations (29%), followed by missense variants (18%) [26]. The project GENIE, led by the American Association for Cancer Research, highlighted how KMT2A is implicated in many diseases, especially in blood cancers such as acute myeloid leukemia (2.49%), T-cell lymphoblastic leukemia (5.63%) and up to 14% in high grade B-cell lymphoma (Figure 5). KMT2A is altered also in 4.65% of malignant solid tumors, such as lung adenocarcinoma, colon adenocarcinoma and bladder urothelial carcinoma (Figure 5). Interestingly, KMT2A is not the only gene of the epigenetic apparatus whose somatic mutations give rise to tumors, especially in blood malignancies. In fact, somatic alterations in other chromatinopathies genes were found in myelodysplastic syndromes (ASXL1, ATRX, DNMT3A, EED, EZH2, KDM6A, KMT2 family genes, PHF6) [50], acute myeloid leukemia (ASXL1, DNMT3A, PHF6) [51], multiple myeloma (KDM6A, KMT2B, KMT2C, WHSC1) [52] and lymphoid malignancies such as acute lymphoblastic leukemia (CREBBP, DNMT3A, EP300, EED, EZH2, PHF6) and diffuse large B-cell lymphoma (CREBBP/EP300, EZH2, KMT2C/D) [52,53].
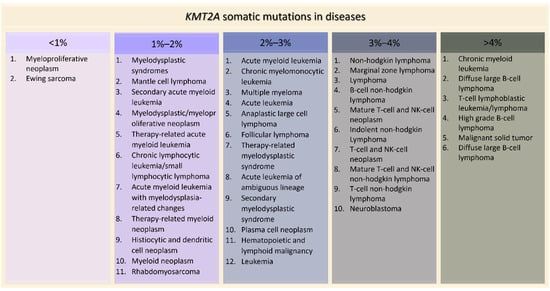
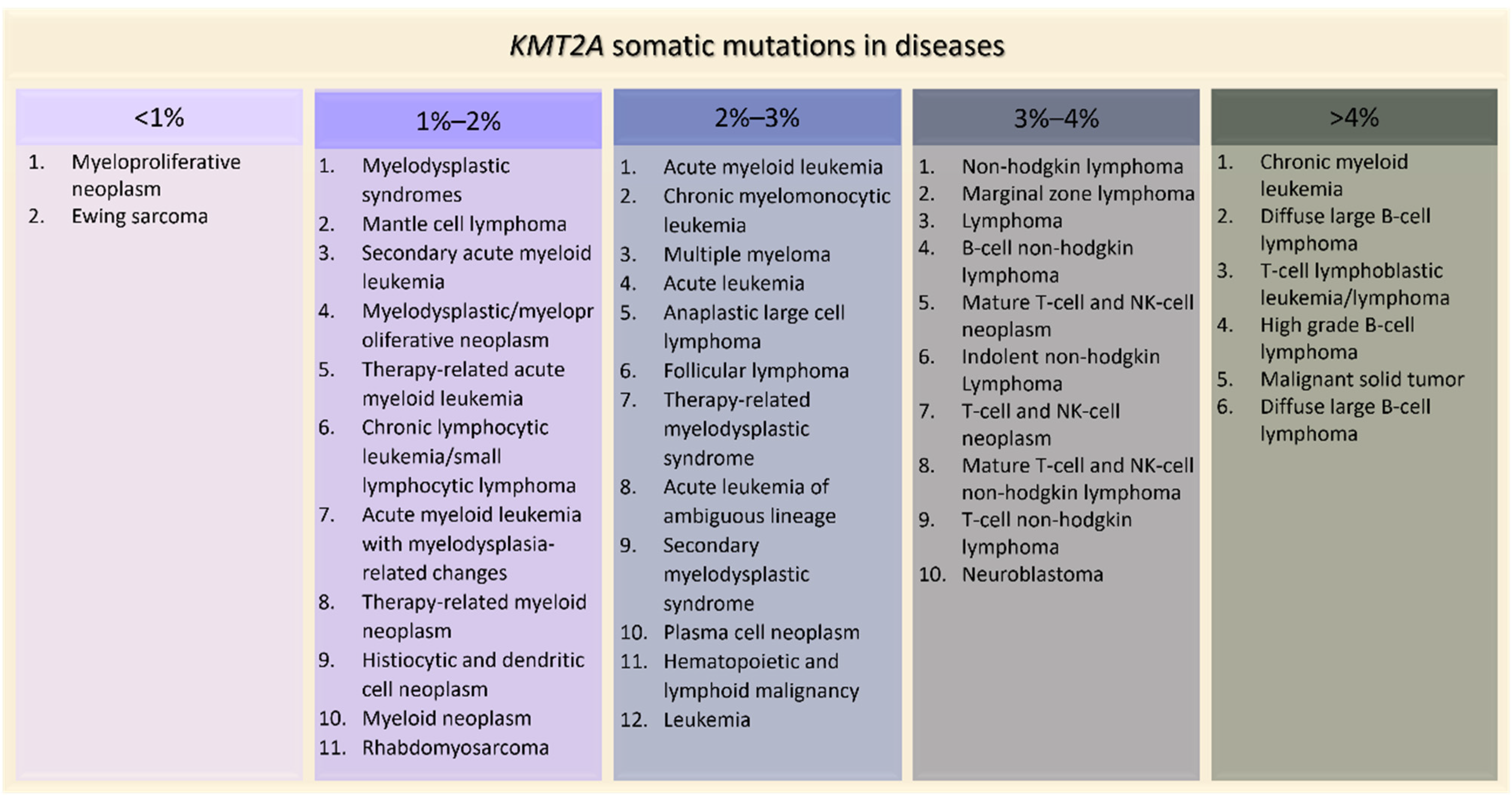
Figure 5.
KMT2A somatic mutations in tumors ordered by percentage of positive cases (AACR Project GENIE).
4. Effects of KMT2A Mutations in Animal Models
KMT2A is an evolutionary conserved gene, involved in several functional process of embryonic development, ranging from hematopoiesis to neurogenesis. Indeed, in 1995, Yu and colleagues showed that the complete disruption of KMT2A was embryonic lethal in mice, and heterozygous animals were anemic and affected by growth delay, hematopoietic anomalies and skeletal malformations [54]. Developmental defects were investigated in Drosophila melanogaster too, where mutations in KMT2A homolog (trx) led to a wide range of homeotic transformations [55]. Interestingly, KMT2A was demonstrated as having an important role in the maintenance of memory Th2 cell function [56] and in hematopoiesis, as its absence caused defects both in self-renewal of murine hematopoietic stem cells and in hematopoietic progenitor cell differentiation in zebrafish [57,58]. In addition, impairments in neural development were observed knocking down Kmt2a in zebrafish, and in murine models Mll1 was identified as a crucial component in memory formation, complex behaviors and synaptic plasticity [59,60,61,62,63].
Thus, KMT2A-depleted animal models recapitulate phenotypes described for patients with both germline and somatic mutations. KMT2A associated syndromes show clinical signs such as ID, behavioral problems, speech and growth delay and peculiar dysmorphisms, while the most frequent tumors enriched in KMT2A mutations are the hematological ones (e.g., B-cell lymphoma, T-cell lymphoblastic leukemia, acute myeloid leukemia), according to neurodevelopmental and hematopoietic defects found in the aforementioned in vivo models.
5. Epigenetic Strategies for Pharmacological Approaches
Targeting the regulators of lysine methylation is an emerging strategy for therapeutic approaches, given the role of chromatin post translational modification in regulating gene expression, and considering that lysine methylation has a pivotal role in this process. Indeed, mutations in one of the components of the epigenetic machinery affect the normal pattern of covalent histone modifications, leading to an incorrect gene expression pattern that may consequently result in tumor evolution. In addition, given the very high specificity of each methyltransferase to its target, the development of drugs directed to those enzymes would have the advantage to minimize the off-target effects [64].
As described above, KMT2A alterations have been reported in several blood cancers such as mixed-lineage, acute lymphoblastic and acute myeloid leukemia [65]. Acute leukemia with rearrangements of the KMT2A gene (KMT2Ar) is associated with a higher risk of relapse and is more resistant to standard therapies. KMT2A exerts its function by forming a core-complex with other proteins [66]; for this reason, the inhibition of KMT2A with its interaction partners, both histone and non-histone proteins, is a promising pharmacological strategy when KMT2A rearrangements are drivers of pathology, such as in leukemia. For example, recent studies have shown that the use of peptidomimetics disrupting the interaction between KMT2A and WDR5 (a member of the above-mentioned core-complex) in murine cell line reduces the expression of target genes responsible for KMT2A-mediated leukemogenesis and inhibits the growth of leukemia cells [67,68].
Similarly, it was demonstrated that the small molecule EPZ-5676 has a modest clinical activity reducing the proliferation of MLL-rearranged cells and inducing apoptosis by targeting the enzymatic core of DOT1L, a H3K79 methyltransferase recruited to fusion partners of KMT2A in disease-linked translocations and required for leukemogenesis [69,70,71,72]. Advances in treating MLL-rearranged leukemia were also achieved by using small molecules to block the KMT2A binding site on Menin, a protein encoded by MEN1 and required for oncogenic transformation, leading to the inhibition of the aberrant leukemogenic transcription program [73,74,75,76,77].
Another pharmacological efficient approach in cancer treatment might be the targeting of pathways deregulated in tumorigenesis. Indeed, the inhibition of glycogen synthase kinase 3 (GSK3) can induce the growth arrest of leukemia cells in KMT2Ar leukemia [78], while targeting the DNA damage response (DDR) pathway can lead to specific synthetic lethality in leukemic cells with MLL-rearrangements [79].
Besides the leukemia treatment, the KMT inhibitors are considered potential drugs for other cancers. In particular, Tazemetostat has been approved in January 2020 for the treatment of a rare tumor, epithelioid sarcoma, and then for follicular lymphoma, sustaining the role of the lysine methylation pathways as potential effective targets for treating various diseases [80].
On the contrary, in genetic disorders related to KMT2A, the altered histone methylation status is mainly attributed to loss of functions mutations or missense mutations involving this gene. For this reason, a possible pharmacological approach could counteract the lack of KMT2A activity.
Altered epigenetic control of gene expression may cause psychosis and other psychiatric diseases, it was demonstrated that the atypical antipsychotic clozapine can induce the methylation of GABAergic gene promoters through Mll1 recruitment in a mouse model of schizophrenia [81,82]. Moreover, a study comparing clozapine-responder and non-responder twins demonstrated that clozapine increases DNA methylation of the MECP2 promoter, leading to its downregulation, and consequently enhancing the expression of genes that are regulated by MeCP2 protein [83]. Similarly, the antidepressant phenelzine and its analogue bizine enhance H3K4me2 status in H460, A549 and MDA-MB-231 cancer cell lines by inhibiting the activity of the histone demethylase LSD1 [84]. Furthermore, tranylcypromine (TCP), another antidepressant, has been demonstrated to specifically inhibit LSD1, and its administration in combination with all-trans-retinoic-acid (ATRA) induces the differentiation of acute promyelocytic leukemia (APL) and acute myeloid leukemia (AML) blasts [85]. Moreover, a phase I/II trial (ClinicalTrials.gov: NCT02261779) have demonstrated that TCP-ATRA combined therapy can be used to treat refractory or relapsed AML patients, even if the required high dosage and the prolonged treatment may cause the onset of several side-effects [86]. For this reason, a selective LSD1 inhibitor, ORY-1001, has been developed using TCP structure. Sub-nanomolar doses of this molecule reduce the proliferation of MLL-translocated leukemic cell lines, both in vitro and in vivo, and display synergistic action with the common anti-leukemic drugs, opening the possibility of a targeted and personalized therapy [87]. A phase I/IIa clinical trial has already evaluated the tolerability, pharmacokinetics and pharmacodynamics of ORY-1001 in relapsed or acute refractory leukemia (EUDRACT no.2013-002447-29) [88].
Interestingly, epigenetic interventions could be either pharmaceutical or nutritional. It is well known that dynamic crosstalk between gut microbiota and the host is present and that it can be modulated by diet. Krautkramer and colleagues reported that microbiota regulates histone methylation and acetylation in different tissues as a diet-dependent process [89] and, notably, a microbiota-dependent epigenetic signature was reported in specific diseases, e.g., inflammatory bowel disease [90]. Indeed, the microbial community within the intestine can produce metabolites such as short-chain fatty acids (SCFAs) with a known role of histone deacetylase (HDAC) inhibitors. These compounds or diets able to increase them were recently used as possible therapeutic approach for several diseases, including drug-resistant epilepsy [91,92], cancer [93], neurodegenerative disease [94], heart failure [95], and diabetes mellitus [96], and their effect was even studied in experimental models of chromatinopathies, i.e., Kabuki syndrome [97] and Rubinstein–Taybi syndrome [98]. Furthermore, bacteria synthetize essential vitamins, fundamental for immune systems, such as B12, but also folate, required for DNA, histone and protein methylation [99,100]. Intriguingly, in a kdm5-deficient Drosophila model, not only an increase in gut H3K4me3 but also the disruption of intestinal barrier together with aberrant immune activation and anomalies in social behavior were observed. All these changes correlated with alterations in gut microbiota composition, which were rescued by probiotic administration [101].
Thus, considering the latest developments on epigenetic intervention, a deepening understanding of microbiota composition of patients with KMT2A mutations could help new therapeutic approaches investigation among the epigenetic treatments.
6. Final Remarks
Epigenetic modifications are fundamental for many biological processes; indeed, alterations of genes with this activity can lead to neurodevelopmental disorders or tumorigenesis, when germinal or somatic mutations respectively occur [102,103]. This is the case of KMT2A, a lysin methyltransferase-coding gene, whose variants are associated with a chromatinopathy (WDSTS) at germinal level or can be found in both blood cancers and solid tumors in regard to malignancies.
Interestingly, due to exome- and genome-wide analyses, patients described above with a defined initial chromatinopathy diagnosis but lacking the molecular one were found to be carriers of pathogenetic variants in the KMT2A gene and could have obtained a clinical re-evaluation. In detail, nearly the totality of patients previously diagnosed with CdLS, CSS, KS and RSTS showed features common to WDSTS, such as ID (11/12), speech delay (7/10), peculiar dysmorphisms affecting eyes (12/12) (i.e., thick eyebrows, synophrys, long eyelashes, ptosis and downslanting/narrow palpebral fissure) and nose (12/12) (i.e., depressed nasal bridge and broad nasal tip), while about half of them shared with WDSTS feeding problems (5/10), hirsutism (6/10) and hypotonia (6/10). Oddly, almost all of these patients displayed features less frequently present in WDSTS, such as dysmorphisms affecting mouth (7/12) (i.e., high arched palate and thin upper vermilion) and anomalies of hands/feet (11/12) (i.e., clinodactyly, brachydactyly, persistent fetal fingerpads and broad halluces). Indeed, mutations in different genes involved in the regulation and maintenance of chromatin state can lead to a clinical overlapping phenotype, suggesting a common affected pathway during embryonic development and the evaluation of an expanded set of genes when investigating the molecular causes for a correct diagnosis of these syndromes.
In addition, somatic mutations in KMT2A have been reported in different tumors, as well as alterations in all KMT2 family genes [104] and in other genes associated to chromatinopathies. Curiously, we observed that germline mutations described in the literature are more frequently nonsense than missense, in contrast to somatic ones. This could be explained by the consequent loss of function mechanism characterizing most of chromatinopathies due to a defective protein production, which strongly impacts on embryonic development.
To conclude, since molecular defects in KMT2A also characterize some types of tumors, and research in the field of epigenetic drugs for malignancies is rapidly evolving [101], a therapeutic approach targeting KMT2A interaction or its pathway could be considered also for chromatinopathies, modulating epigenetic dysfunction with pharmaceutical products or diet-based interventions.
Author Contributions
C.G. and V.M. conceived the manuscript. S.C. and E.D.F. wrote the manuscript. A.V., D.M. and F.G. reviewed clinical information. C.B., E.L., E.O. and E.B. wrote sections of the manuscript. A.L., C.P., P.G., E.A.C. and S.A. read and edited the manuscript. C.G. and V.M. reviewed and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful to the following funding sources: Intramural funding—Dipartimento DISS, Linea Incentivo alla Ricerca, Università degli Studi di Milano (to E.B., F.G., C.G., E.L., V.M. and A.V.); Translational Medicine Ph.D. scholarship—Università degli Studi di Milano (to E.D.F. and C.P.); Molecular and Translational Medicine Ph.D. scholarship—Università degli Studi di Milano (to P.G.); and the “Aldo Ravelli” Center for Neurotechnology and Experimental Brain Therapeutics—Università degli Studi di Milano (to C.G., A.L. and V.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, B.A.; Hake, S.B.; Diaz, R.L.; Kauer, M.; Morris, S.A.; Recht, J.; Shabanowitz, J.; Mishra, N.; Strahl, B.D.; Allis, C.D.; et al. Organismal Differences in Post-Translational Modifications in Histones H3 and H4. J. Biol. Chem. 2007, 282, 7641–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojer, P.; Zhang, J.; Yonezawa, M.; Schmidt, A.; Zheng, H.; Jenuwein, T.; Reinberg, D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-Containing JMJD2/KDM4 Proteins. J. Biol. Chem. 2009, 284, 8395–8405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daujat, S.; Weiss, T.; Mohn, F.; Lange, U.C.; Ziegler-Birling, C.; Zeissler, U.; Lappe, M.; Schübeler, D.; Torres-Padilla, M.E.; Schneider, R. H3K64 Trimethylation Marks Heterochromatin and Is Dynamically Remodeled during Developmental Reprogramming. Nat. Struct. Mol. Biol. 2009, 16, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Hergeth, S.; Zeissler, U.; Izzo, A.; Tropberger, P.; Zee, B.M.; Dundr, M.; Garcia, B.A.; Daujat, S.; Schneider, R. Histone H1 Variant-Specific Lysine Methylation by G9a/KMT1C and Glp1/KMT1D. Epigenet. Chromatin 2010, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Paredes, M.; Esteller, M. Cancer Epigenetics Reaches Mainstream Oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.F.; McDevitt, P.; Sinnamon, R.; Le, B.C.; Mas, G.; et al. Smyd3 Regulates Cancer Cell Phenotypes and Catalyzes Histone H4 Lysine 5 Methylation. Epigenetics 2012, 7, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husmann, D.; Gozani, O. Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, S.; Chen, T.Y.; Lei, M.; Dhar, S.S.; Ho, J.C.; Dong, A.; Loppnau, P.; Li, Y.; Lee, M.G.; et al. Structural Insights into Trans-Histone Regulation of H3K4 Methylation by Unique Histone H4 Binding of MLL3/4. Nat. Commun. 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lee, Y.T.; Zhou, Y.; Huang, Y. Targeting Epigenetic Regulatory Machinery to Overcome Cancer Therapy Resistance. Semin. Cancer Biol. 2021, in press. [CrossRef]
- Zhang, T.; Zhang, W.; Liu, L.; Chen, Y. Simultaneous Detection of Site-Specific Histone Methylations and Acetylation Assisted by Single Template Oriented Molecularly Imprinted Polymers. Analyst 2020, 145, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Di Nisio, E.; Lupo, G.; Licursi, V.; Negri, R. The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation. Front. Genet. 2021, 12, 482. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.P.; Habib, F.; Sharma, R.; Gadewal, N.; Gupta, S.; Galande, S. HIstome—A Relational Knowledgebase of Human Histone Proteins and Histone Modifying Enzymes. Nucleic Acids Res. 2012, 40, D337–D342. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Tipping the Balance of Chromatin States. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [Green Version]
- Bjornsson, H.T. The Mendelian Disorders of the Epigenetic Machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Postnatal Malleability and Therapeutic Prospects. Hum. Mol. Genet. 2019, 28, R254–R264. [Google Scholar] [CrossRef]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.L. MLL: A Histone Methyltransferase Disrupted in Leukemia. Trends Mol. Med. 2004, 10, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Southall, S.M.; Wong, P.S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33, 181–191. [Google Scholar] [CrossRef]
- Wiedemann, H.R.; Kunze, J.; Grosse, F.R.; Dibbern, H. A Syndrome of Abnormal Facies, Short Stature, and Psychomotor Retardation. In Atlas of Clinical Syndromes: A Visual Aid to Diagnosis for Clinicians and Practicing Physicians, 2nd ed.; Wolfe Publishing Ltd.: London, UK, 1989; pp. 198–199. [Google Scholar]
- Miyake, N.; Tsurusaki, Y.; Koshimizu, E.; Okamoto, N.; Kosho, T.; Brown, N.J.; Tan, T.Y.; Yap, P.J.J.; Suzumura, H.; Tanaka, T.; et al. Delineation of Clinical Features in Wiedemann-Steiner Syndrome Caused by KMT2A Mutations. Clin. Genet. 2016, 89, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Rodriguez-Buritica, D.F.; Northrup, H. Wiedemann-Steiner Syndrome: Novel Pathogenic Variant and Review of Literature. Eur. J. Med. Genet. 2017, 60, 285–288. [Google Scholar] [CrossRef] [PubMed]
- di Fede, E.; Massa, V.; Augello, B.; Squeo, G.; Scarano, E.; Perri, A.M.; Fischetto, R.; Causio, F.A.; Zampino, G.; Piccione, M.; et al. Expanding the Phenotype Associated to KMT2A Variants: Overlapping Clinical Signs between Wiedemann–Steiner and Rubinstein–Taybi Syndromes. Eur. J. Hum. Genet. 2021, 29, 88–98. [Google Scholar] [CrossRef]
- Masciadri, M.; Ficcadenti, A.; Milani, D.; Cogliati, F.; Divizia, M.T.; Larizza, L.; Russo, S. Recurrence and Familial Inheritance of Intronic NIPBL Pathogenic Variant Associated With Mild CdLS. Front. Neurol. 2018, 9, 967. [Google Scholar] [CrossRef]
- Milani, D.; Manzoni, F.M.P.; Pezzani, L.; Ajmone, P.; Gervasini, C.; Menni, F.; Esposito, S. Rubinstein-Taybi Syndrome: Clinical Features, Genetic Basis, Diagnosis, and Management. Ital. J. Pediatr. 2015, 41, 4. [Google Scholar] [CrossRef] [Green Version]
- Bramswig, N.C.; Lüdecke, H.J.; Alanay, Y.; Albrecht, B.; Barthelmie, A.; Boduroglu, K.; Braunholz, D.; Caliebe, A.; Chrzanowska, K.H.; Czeschik, J.C.; et al. Exome Sequencing Unravels Unexpected Differential Diagnoses in Individuals with the Tentative Diagnosis of Coffin–Siris and Nicolaides–Baraitser Syndromes. Hum. Genet. 2015, 134, 553–568. [Google Scholar] [CrossRef]
- Schrier Vergano, S.; Santen, G.; Wieczorek, D.; Wollnik, B.; Matsumoto, N.; Deardoff, M.A. Coffin-Siris Syndrome. In GeneReviews [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; NCBI: Seattle, WA, USA, 2013. [Google Scholar]
- Sobreira, N.; Brucato, M.; Zhang, L.; Ladd-Acosta, C.; Ongaco, C.; Romm, J.; Doheny, K.F.; Mingroni-Netto, R.C.; Bertola, D.; Kim, C.A.; et al. Patients with a Kabuki Syndrome Phenotype Demonstrate DNA Methylation Abnormalities. Eur. J. Hum. Genet. 2017, 25, 1335–1344. [Google Scholar] [CrossRef]
- Wang, Y.R.; Xu, N.X.; Wang, J.; Wang, X.M. Kabuki Syndrome: Review of the Clinical Features, Diagnosis and Epigenetic Mechanisms. World J. Pediatr. WJP 2019, 15, 528–535. [Google Scholar] [CrossRef]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 Lysine 4 Methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.I.; Jespersgaard, C.; Brøndum-Nielsen, K.; Bisgaard, A.M.; Tümer, Z. Cornelia de Lange Syndrome. Clin. Genet. 2015, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Pehlivan, D.; Karaca, E.; Patel, N.; Charng, W.-L.; Gambin, T.; Gonzaga-Jauregui, C.; Sutton, V.R.; Yesil, G.; Bozdogan, S.T.; et al. Global Transcriptional Disturbances Underlie Cornelia de Lange Syndrome and Related Phenotypes. J. Clin. Investig. 2015, 125, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Teresa-Rodrigo, M.E.; Pozojevic, J.; Ruiz Gil, S.; Bader, I.; Braunholz, D.; Bramswig, N.C.; Gervasini, C.; Larizza, L.; Pfeiffer, L.; et al. Mutations in Chromatin Regulators Functionally Link Cornelia de Lange Syndrome and Clinically Overlapping Phenotypes. Hum. Genet. 2017, 136, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Negri, G.; Magini, P.; Milani, D.; Crippa, M.; Biamino, E.; Piccione, M.; Sotgiu, S.; Perrìa, C.; Vitiello, G.; Frontali, M.; et al. Exploring by Whole Exome Sequencing Patients with Initial Diagnosis of Rubinstein–Taybi Syndrome: The Interconnections of Epigenetic Machinery Disorders. Hum. Genet. 2019, 138, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, J.H.; Taybi, H. Broad Thumbs and Toes and Facial Abnormalities. A Possible Mental Retardation Syndrome. Am. J. Dis. Child. 1963, 105, 588–608. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C.M. Rubinstein–Taybi Syndrome. Eur. J. Hum. Genet. 2006, 14, 981–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholdi, D.; Roelfsema, J.H.; Papadia, F.; Breuning, M.H.; Niedrist, D.; Hennekam, R.C.; Schinzel, A.; Peters, D.J.M. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Delineation of the Phenotype of the First Patients Carrying Mutations in EP300. J. Med. Genet. 2007, 44, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Fergelot, P.; Van Belzen, M.; Van Gils, J.; Afenjar, A.; Armour, C.M.; Arveiler, B.; Beets, L.; Burglen, L.; Busa, T.; Collet, M.; et al. Phenotype and Genotype in 52 Patients with Rubinstein–Taybi Syndrome Caused by EP300 Mutations. Am. J. Med. Genet. Part A 2016, 170, 3069–3082. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene Dosage-Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator P300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Oike, Y.; Hata, A.; Mamiya, T.; Kaname, T.; Noda, Y.; Suzuki, M.; Yasue, H.; Nabeshima, T.; Araki, K.; Yamamura, K. Truncated CBP Protein Leads to Classical Rubinstein-Taybi Syndrome Phenotypes in Mice: Implications for a Dominant-Negative Mechanism. Hum. Mol. Genet. 1999, 8, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, H.M.; La Thangue, N.B. P300/CBP Proteins: HATs for Transcriptional Bridges and Scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Tillhon, M.; Cazzalini, O.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. P300/CBP Acetyl Transferases Interact with and Acetylate the Nucleotide Excision Repair Factor XPG. DNA Repair 2012, 11, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Dutto, I.; Scalera, C.; Prosperi, E. CREBBP and P300 Lysine Acetyl Transferases in the DNA Damage Response. Cell. Mol. Life Sci. 2018, 75, 1325–1338. [Google Scholar] [CrossRef]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; De Braekeleer, E.; De Braekeleer, M.; et al. New Insights to the MLL Recombinome of Acute Leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef]
- Sweeney, S.M.; Cerami, E.; Baras, A.; Pugh, T.J.; Schultz, N.; Stricker, T.; Lindsay, J.; Del Vecchio Fitz, C.; Kumari, P.; Micheel, C.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic Cancer Genetics at High-Resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Kishtagari, A.; Levine, R.L. The Role of Somatic Mutations in Acute Myeloid Leukemia Pathogenesis. Cold Spring Harb. Perspect. Med. 2021, 11, a034975. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Genome Sequencing of Lymphoid Malignancies. Blood 2013, 122, 3899–3907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Cai, K.; Xu, P.P.; Wang, L.; Huang, C.X.; Fang, Y.; Cheng, S.; Sun, X.J.; Liu, F.; Huang, J.Y.; et al. CREBBP/EP300 Mutations Promoted Tumor Progression in Diffuse Large B-Cell Lymphoma through Altering Tumor-Associated Macrophage Polarization via FBXW7-NOTCH-CCL2/CSF1 Axis. Signal Transduct. Target. Ther. 2021, 6, 10. [Google Scholar] [CrossRef]
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.J.; Korsmeyer, S.J. Altered HOX Expression and Segmental Identity in Mll-Mutant Mice. Nature 1995, 378, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Katsani, K.R.; Arredondo, J.J.; Kal, A.J.; Verrijzer, C.P. A Homeotic Mutation in the Trithorax SET Domain Impedes Histone Binding. Genes Dev. 2001, 15, 2197–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Hirahara, K.; Shinnakasu, R.; Hosokawa, H.; Norikane, S.; Kimura, M.Y.; Hasegawa, A.; Nakayama, T. Crucial Role of MLL for the Maintenance of Memory T Helper Type 2 Cell Responses. Immunity 2006, 24, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, K.A.; Hiew, S.Y.L.; Hadjur, S.; Veiga-Fernandes, H.; Menzel, U.; Price, A.J.; Kioussis, D.; Williams, O.; Brady, H.J.M. Mll Has a Critical Role in Fetal and Adult Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2007, 1, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Hu, B.; Liu, J.X.; Feng, X.; Xiao, W. Zebrafish Mll Gene Is Essential for Hematopoiesis. J. Biol. Chem. 2011, 286, 33345–33357. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.C.; Shih, H.Y.; Lin, S.J.; Chiu, C.C.; Ma, T.L.; Yeh, T.H.; Cheng, Y.C. The Epigenetic Factor Kmt2a/Mll1 Regulates Neural Progenitor Proliferation and Neuronal and Glial Differentiation. Dev. Neurobiol. 2015, 75, 452–462. [Google Scholar] [CrossRef]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone Methylation Regulates Memory Formation. J. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Kerimoglu, C.; Sakib, M.S.; Jain, G.; Benito, E.; Burkhardt, S.; Capece, V.; Kaurani, L.; Halder, R.; Agís-Balboa, R.C.; Stilling, R.; et al. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep. 2017, 20, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovcevski, M.; Ruan, H.; Shen, E.Y.; Dincer, A.; Javidfar, B.; Ma, Q.; Peter, C.J.; Cheung, I.; Mitchell, A.C.; Jiang, Y.; et al. Neuronal Kmt2a/Mll1 Histone Methyltransferase Is Essential for Prefrontal Synaptic Plasticity and Working Memory. J. Neurosci. 2015, 35, 5097–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, E.Y.; Jiang, Y.; Javidfar, B.; Kassim, B.; Loh, Y.H.E.; Ma, Q.; Mitchell, A.C.; Pothula, V.; Stewart, A.F.; Ernst, P.; et al. Neuronal Deletion of Kmt2a/Mll1 Histone Methyltransferase in Ventral Striatum Is Associated with Defective Spike-Timing-Dependent Striatal Synaptic Plasticity, Altered Response to Dopaminergic Drugs, and Increased Anxiety. Neuropsychopharmacology 2016, 41, 3103–3113. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.; Ümit Kaniskan, H.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting Writers of Protein Lysine Methylation to Treat Disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Tenney, K.; Shilatifard, A. A COMPASS in the Voyage of Defining the Role of Trithorax/MLL-Containing Complexes: Linking Leukemogensis to Covalent Modifications of Chromatin. J. Cell. Biochem. 2005, 95, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Yao, T.; Cao, M.; Zhu, G.; Li, Y.; Yuan, G.; Chen, Y.; Lei, M.; Huang, J. Structural Basis of Nucleosome Recognition and Modification by MLL Methyltransferases. Nature 2019, 573, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Townsend, E.C.; Cao, F.; Chen, Y.; Bernard, D.; Liu, L.; Lei, M.; Dou, Y.; Wang, S. High-Affinity, Small-Molecule Peptidomimetic Inhibitors of MLL1/WDR5 Protein-Protein Interaction. J. Am. Chem. Soc. 2013, 135, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y.; Ouillette, P.; Zhu, J.; et al. Targeting MLL1 H3K4 Methyltransferase Activity in Mixed-Lineage Leukemia. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent Inhibition of DOT1L as Treatment of MLL-Fusion Leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L Inhibitor Pinometostat Reduces H3K79 Methylation and Has Modest Clinical Activity in Adult Acute Leukemia. Blood 2018, 131, 2662–2669. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia in Vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Hong, X.Y.; Zhu, L.Y.; Zhang, L.; Qiu, H.; Zhang, Y.Y.; Yuan, M.C.; Zhao, X.L.; Zheng, Q.F.; Jin, G.H. Abnormal Expression of Menin Predicts the Pathogenesis and Poor Prognosis of Adult Gliomas. Cancer Gene Ther. 2020, 27, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic Implications of Menin Inhibition in Acute Leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef]
- Grieselhuber, N.R.; Mims, A.S. Novel Targeted Therapeutics in Acute Myeloid Leukemia: An Embarrassment of Riches. Curr. Hematol. Malig. Rep. 2021, 16, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.P.; Cleary, M.L. Glycogen Synthase Kinase 3 in MLL Leukaemia Maintenance and Targeted Therapy. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef] [Green Version]
- Tsakaneli, A.; Williams, O. Drug Repurposing for Targeting Acute Leukemia With KMT2A ( MLL)-Gene Rearrangements. Front. Pharmacol. 2021, 12, 2513. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Baylin, S.B. Epigenetic Therapy for Epithelioid Sarcoma. Cell 2020, 181, 211. [Google Scholar] [CrossRef]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The Histone H3K4 Demethylase SMCX Links REST Target Genes to X-Linked Mental Retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Matevossian, A.; Whittle, C.; Se, Y.K.; Schumacher, A.; Baker, S.P.; Akbarian, S. Prefrontal Dysfunction in Schizophrenia Involves Mixed-Lineage Leukemia 1-Regulated Histone Methylation at GABAergic Gene Promoters. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 11254–11262. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, M.; Nakazawa, T.; Kinoshita, M.; Yamamori, H.; Yasuda, Y.; Fujimoto, M.; Hashimoto, R.; Numata, S. Methylation Analysis in Monozygotic Twins With Treatment-Resistant Schizophrenia and Discordant Responses to Clozapine. Front. Psychiatry 2021, 12, 1610. [Google Scholar] [CrossRef] [PubMed]
- Prusevich, P.; Kalin, J.H.; Ming, S.A.; Basso, M.; Givens, J.; Li, X.; Hu, J.; Taylor, M.S.; Cieniewicz, A.M.; Hsiao, P.Y.; et al. A Selective Phenelzine Analogue Inhibitor of Histone Demethylase LSD1. ACS Chem. Biol. 2014, 9, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) Demethylase Reactivates the All-Trans-Retinoic Acid Differentiation Pathway in Acute Myeloid Leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Wass, M.; Göllner, S.; Besenbeck, B.; Schlenk, R.F.; Mundmann, P.; Göthert, J.R.; Noppeney, R.; Schliemann, C.; Mikesch, J.H.; Lenz, G.; et al. A Proof of Concept Phase I/II Pilot Trial of LSD1 Inhibition by Tranylcypromine Combined with ATRA in Refractory/Relapsed AML Patients Not Eligible for Intensive Therapy. Leukemia 2021, 35, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamero, O.; Montesinos, P.; Willekens, C.; Pérez-Simón, J.A.; Pigneux, A.; Récher, C.; Popat, R.; Carpio, C.; Molinero, C.; Mascaró, C.; et al. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.; Yang, L.; Pei, Z. Gut Microbiota, Fusobacteria, and Colorectal Cancer. Diseases 2018, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Vining, E.P.G. Gaining a Perspective on Childhood Seizures. N. Engl. J. Med. 1998, 338, 1916–1918. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Zuckermann, A.M.E.; Williams, S.; Close, A.J.; Cano-Jaimez, M.; McEvoy, J.P.; Spencer, J.; Walker, M.C.; Williams, R.S.B. Seizure Control by Derivatives of Medium Chain Fatty Acids Associated with the Ketogenic Diet Show Novel Branching-Point Structure for Enhanced Potency. J. Pharmacol. Exp. Ther. 2015, 352, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Shingler, E.; Perry, R.; Mitchell, A.; England, C.; Perks, C.; Herbert, G.; Ness, A.; Atkinson, C. Dietary Restriction during the Treatment of Cancer: Results of a Systematic Scoping Review. BMC Cancer 2019, 19, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosinski, C.; Jornayvaz, F.R. Effects of Ketogenic Diets on Cardiovascular Risk Factors: Evidence from Animal and Human Studies. Nutrients 2017, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Westman, E.C.; Vernon, M.C. Has Carbohydrate-Restriction Been Forgotten as a Treatment for Diabetes Mellitus? A Perspective on the ACCORD Study Design. Nutr. Metab. 2008, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, J.S.; Pilarowski, G.O.; Carosso, G.A.; Zhang, L.; Huso, D.L.; Goff, L.A.; Vernon, H.J.; Hansen, K.D.; Bjornsson, H.T. A Ketogenic Diet Rescues Hippocampal Memory Defects in a Mouse Model of Kabuki Syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fede, E.; Ottaviano, E.; Grazioli, P.; Ceccarani, C.; Galeone, A.; Parodi, C.; Colombo, E.A.; Bassanini, G.; Fazio, G.; Severgnini, M.; et al. Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein–Taybi Syndrome. Int. J. Mol. Sci. 2021, 22, 3621. [Google Scholar] [CrossRef] [PubMed]
- Mckay, J.A.; Mathers, J.C. Diet Induced Epigenetic Changes and Their Implications for Health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA Methylation: A Review of Molecular Mechanisms and the Evidence for Folate’s Role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.H.; Liu, Y.W.; Wu, C.C.; Wang, S.; Tsai, Y.C. Psychobiotics in Mental Health, Neurodegenerative and Neurodevelopmental Disorders. J. Food Drug Anal. 2019, 27, 632–648. [Google Scholar] [CrossRef] [Green Version]
- Jakovcevski, M.; Akbarian, S. Epigenetic Mechanisms in Neurological Disease. Nat. Med. 2012, 18, 1194–1204. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagan, R.J.; Dingwall, A.K. COMPASS Ascending: Emerging Clues Regarding the Roles of MLL3/KMT2C and MLL2/KMT2D Proteins in Cancer. Cancer Lett. 2019, 458, 56–65. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).