
Implications of Dosage Deficiencies in CTCF and Cohesin on Genome Organization, Gene Expression, and Human Neurodevelopment


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Main
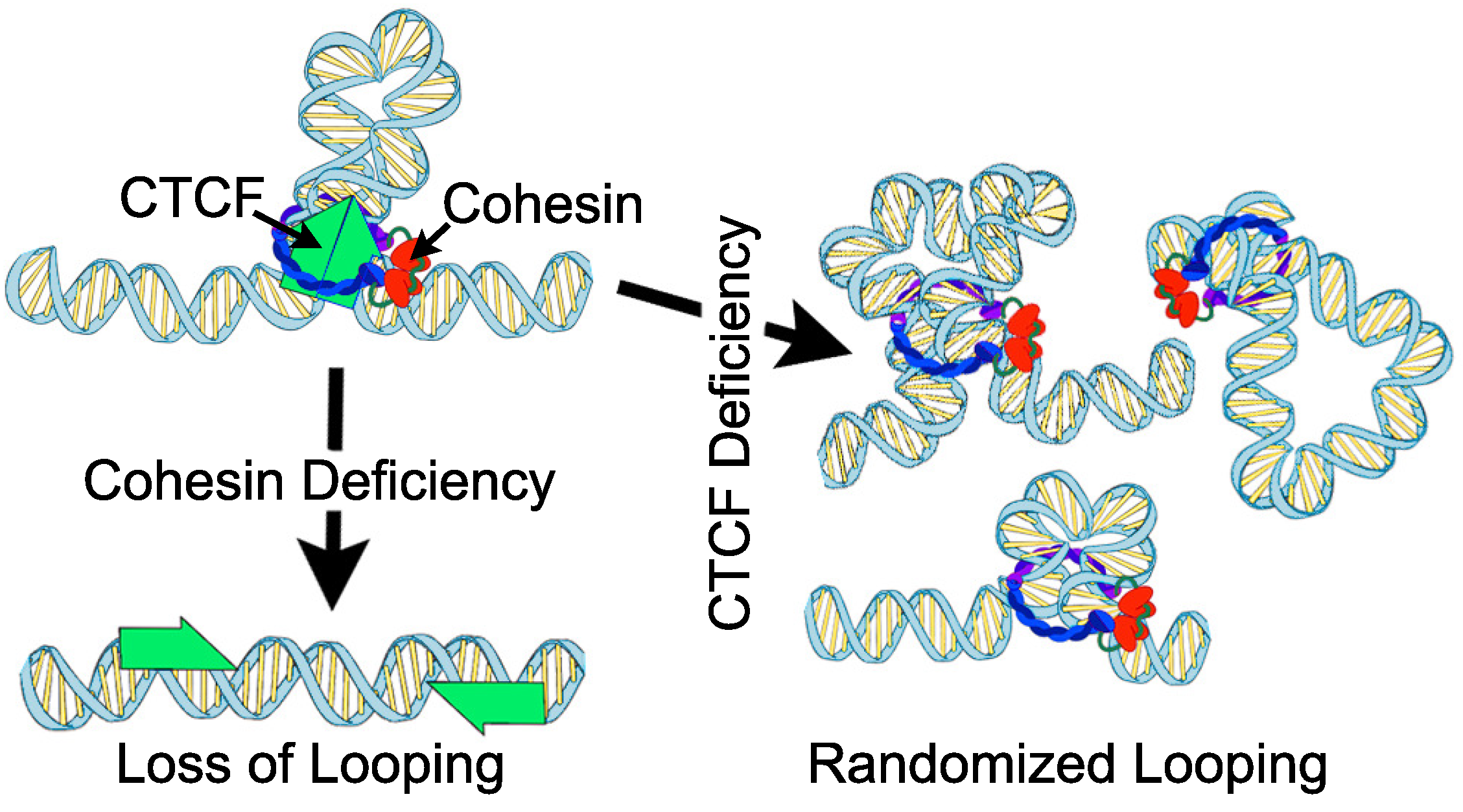
2.1. Chromatin Organization and Gene Expression upon CTCF or Cohesin Depletion
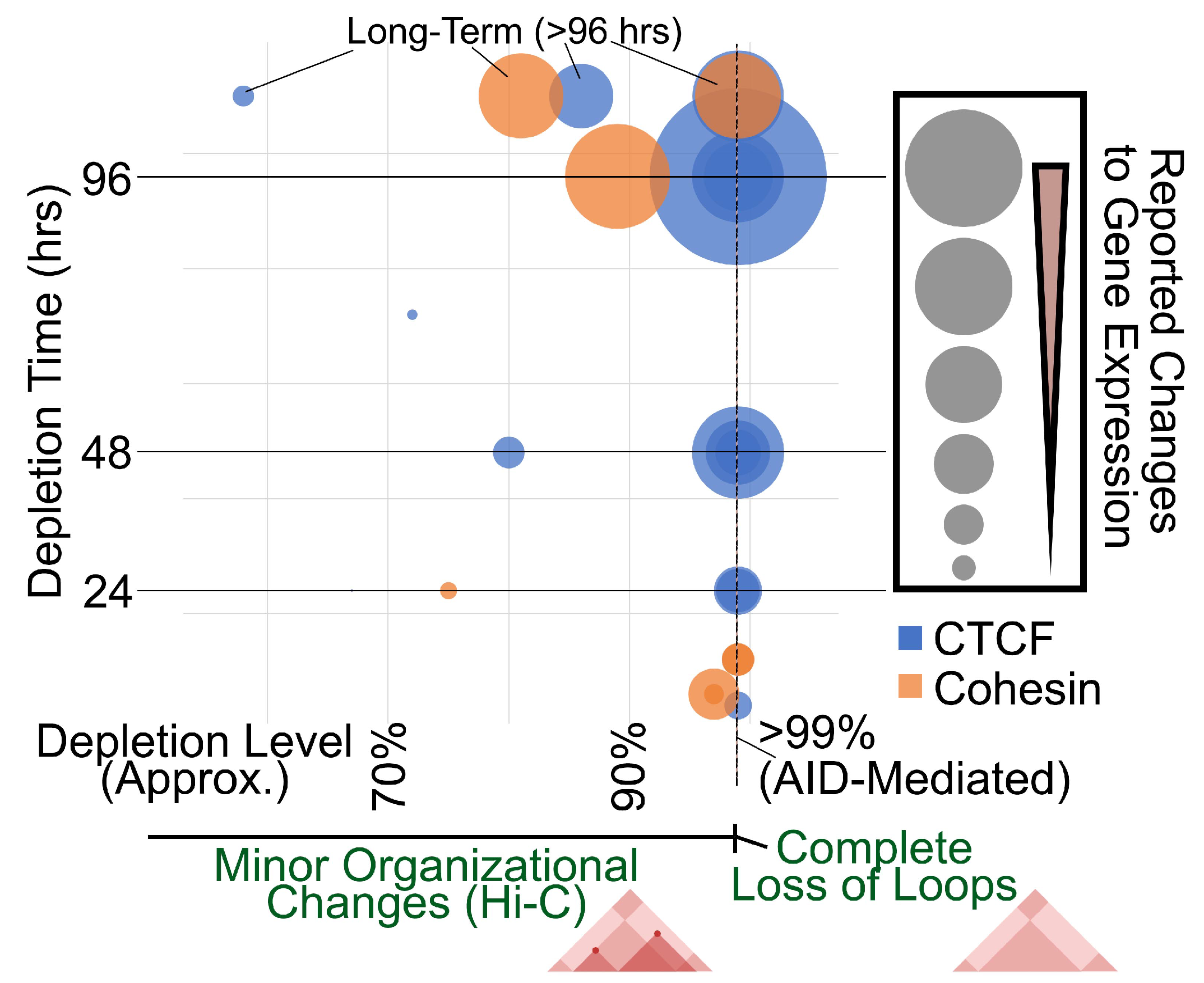
2.2. Evidence of Dosage Sensitivity to CTCF and Cohesin Depletion
2.3. Deletion of CTCF and Cohesin in Mouse Models
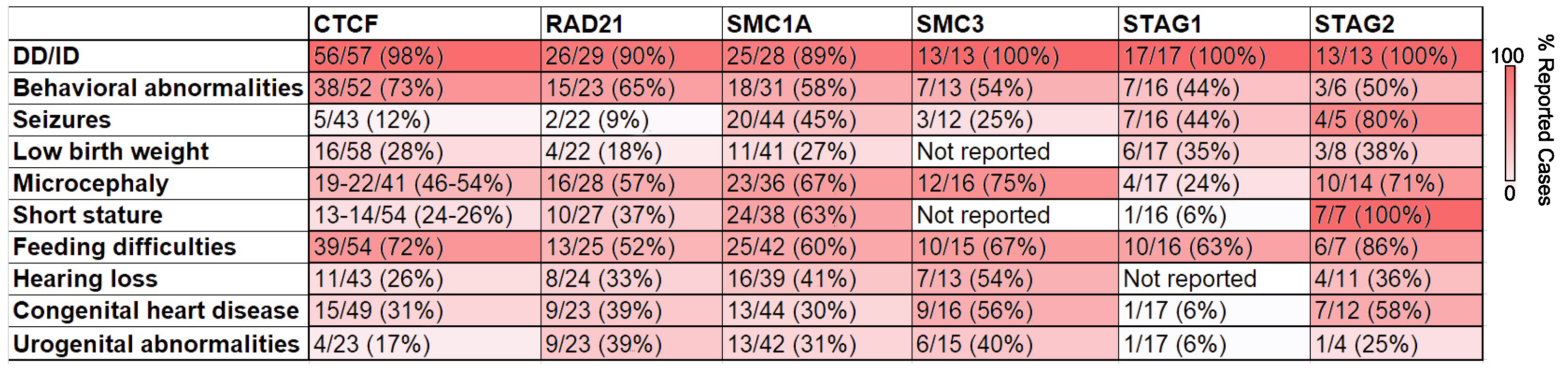
2.4. Implications of Germline Variants of CTCF and Cohesin in Humans
3. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Phipps, J.; Dubrana, K. DNA Repair in Space and Time: Safeguarding the Genome with the Cohesin Complex. Genes 2022, 13, 198. [Google Scholar] [CrossRef] [PubMed]
- Yatskevich, S.; Rhodes, J.; Nasmyth, K. Organization of Chromosomal DNA by SMC Complexes. Annu. Rev. Genet. 2019, 53, 445–482. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef] [Green Version]
- Nichols, M.H.; Corces, V.G. A CTCF Code for 3D Genome Architecture. Cell 2015, 162, 703–705. [Google Scholar] [CrossRef] [Green Version]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Nasmyth, K. Disseminating the genome: Joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu. Rev. Genet. 2001, 35, 673–745. [Google Scholar] [CrossRef] [Green Version]
- Alipour, E.; Marko, J.F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 2012, 40, 11202–11212. [Google Scholar] [CrossRef] [Green Version]
- Stigler, J.; Çamdere, G.Ö.; Koshland, D.E.; Greene, E.C. Single-Molecule Imaging Reveals a Collapsed Conformational State for DNA-Bound Cohesin. Cell Rep. 2016, 15, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, I.F.; Goetz, D.; Zaczek, M.P.; Molodtsov, M.I.; Huis In ’t Veld, P.J.; Weissmann, F.; Litos, G.; Cisneros, D.A.; Ocampo-Hafalla, M.; Ladurner, R.; et al. Rapid movement and transcriptional re-localization of human cohesin on DNA. Embo J. 2016, 35, 2671–2685. [Google Scholar] [CrossRef] [PubMed]
- Ganji, M.; Shaltiel, I.A.; Bisht, S.; Kim, E.; Kalichava, A.; Haering, C.H.; Dekker, C. Real-time imaging of DNA loop extrusion by condensin. Science 2018, 360, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Kerssemakers, J.; Shaltiel, I.A.; Haering, C.H.; Dekker, C. DNA-loop extruding condensin complexes can traverse one another. Nature 2020, 579, 438–442. [Google Scholar] [CrossRef]
- Bauer, B.W.; Davidson, I.F.; Canena, D.; Wutz, G.; Tang, W.; Litos, G.; Horn, S.; Hinterdorfer, P.; Peters, J.M. Cohesin mediates DNA loop extrusion by a “swing and clamp” mechanism. Cell 2021, 184, 5448–5464 e22. [Google Scholar] [CrossRef]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Chronis, C.; Kronenberg, M.; Chen, X.F.; Su, T.; Lay, F.D.; Plath, K.; Kurdistani, S.K.; Carey, M.F. Promoter-Enhancer Communication Occurs Primarily within Insulated Neighborhoods. Mol. Cell 2019, 73, 250–263.e5. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Huang, S.C.; Glenn St. Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bochkov, I.D.; Huang, X.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Williamson, I.; Kane, L.; Devenney, P.S.; Flyamer, I.M.; Anderson, E.; Kilanowski, F.; Hill, R.E.; Bickmore, W.A.; Lettice, L.A. Developmentally regulated Shh expression is robust to TAD perturbations. Development 2019, 146, dev179523. [Google Scholar] [CrossRef] [Green Version]
- Banerji, R.; Skibbens, R.V.; Iovine, M.K. How many roads lead to cohesinopathies? Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2017, 246, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruijn, S.E.; Fiorentino, A.; Ottaviani, D.; Fanucchi, S.; Melo, U.S.; Corral-Serrano, J.C.; Mulders, T.; Georgiou, M.; Rivolta, C.; Pontikos, N.; et al. Structural Variants Create New Topological-Associated Domains and Ectopic Retinal Enhancer-Gene Contact in Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2020, 107, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schopflin, R.; Kraft, K.; Kempfer, R.; Jerkovic, I.; Chan, W.L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, M.; Brancati, F.; Krawitz, P.M.; Robinson, P.N.; Ibrahim, D.M.; Franke, M.; Hecht, J.; Lohan, S.; Dathe, K.; Nardone, A.M.; et al. Homeotic arm-to-leg transformation associated with genomic rearrangements at the PITX1 locus. Am. J. Hum. Genet. 2012, 91, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobanenkov, V.V.; Nicolas, R.H.; Adler, V.V.; Paterson, H.; Klenova, E.M.; Polotskaja, A.V.; Goodwin, G.H. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5’-flanking sequence of the chicken c-myc gene. Oncogene 1990, 5, 1743–1753. [Google Scholar]
- Yao, H.; Brick, K.; Evrard, Y.; Xiao, T.; Camerini-Otero, R.D.; Felsenfeld, G. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev. 2010, 24, 2543–2555. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Filippova, G.; Loukinov, D.; Pugacheva, E.; Chen, Q.; Smith, S.T.; Munhall, A.; Grewe, B.; Bartkuhn, M.; Arnold, R.; et al. CTCF is conserved from Drosophila to humans and confers enhancer blocking of the Fab-8 insulator. EMBO Rep. 2005, 6, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, A.; Mohana, G.; Dorier, J.; Ozdemir, I.; Omer, A.; Cousin, P.; Semenova, A.; Taschner, M.; Dergai, O.; Marzetta, F.; et al. CTCF loss has limited effects on global genome architecture in Drosophila despite critical regulatory functions. Nat. Commun. 2021, 12, 1011. [Google Scholar] [CrossRef]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.; Ye, Z.; Kolovos, P.; Brouwer, R.W.; van de Corput, M.P.; van de Werken, H.J.; Knoch, T.A.; van, I.W.F.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [Green Version]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofueva, S.; Yaffe, E.; Chan, W.C.; Georgopoulou, D.; Vietri Rudan, M.; Mira-Bontenbal, H.; Pollard, S.M.; Schroth, G.P.; Tanay, A.; Hadjur, S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013, 32, 3119–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wutz, G.; Varnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Ishii, H.; Xiong, X.; Bianco, S.; Meitinger, F.; Hu, R.; Hocker, J.D.; Conte, M.; Gorkin, D.; Yu, M.; et al. Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat. Struct. Mol. Biol. 2021, 28, 152–161. [Google Scholar] [CrossRef]
- Casa, V.; Moronta Gines, M.; Gade Gusmao, E.; Slotman, J.A.; Zirkel, A.; Josipovic, N.; Oole, E.; van, I.W.F.J.; Houtsmuller, A.B.; Papantonis, A.; et al. Redundant and specific roles of cohesin STAG subunits in chromatin looping and transcriptional control. Genome Res. 2020, 30, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Wutz, G.; Ladurner, R.; St Hilaire, B.G.; Stocsits, R.R.; Nagasaka, K.; Pignard, B.; Sanborn, A.; Tang, W.; Varnai, C.; Ivanov, M.P.; et al. ESCO1 and CTCF enable formation of long chromatin loops by protecting cohesin(STAG1) from WAPL. Elife 2020, 9, e52091. [Google Scholar] [CrossRef]
- Luan, J.; Xiang, G.; Gomez-Garcia, P.A.; Tome, J.M.; Zhang, Z.; Vermunt, M.W.; Zhang, H.; Huang, A.; Keller, C.A.; Giardine, B.M.; et al. Distinct properties and functions of CTCF revealed by a rapidly inducible degron system. Cell Rep. 2021, 34, 108783. [Google Scholar] [CrossRef]
- Khoury, A.; Achinger-Kawecka, J.; Bert, S.A.; Smith, G.C.; French, H.J.; Luu, P.L.; Peters, T.J.; Du, Q.; Parry, A.J.; Valdes-Mora, F.; et al. Constitutively bound CTCF sites maintain 3D chromatin architecture and long-range epigenetically regulated domains. Nat. Commun. 2020, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Hyle, J.; Zhang, Y.; Wright, S.; Xu, B.; Shao, Y.; Easton, J.; Tian, L.; Feng, R.; Xu, P.; Li, C. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer-promoter looping. Nucleic Acids Res. 2019, 47, 6699–6713. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.D.P.; Feldmann, A.; Hernández-Rodríguez, B.; Díaz, N.; Brown, J.M.; Fursova, N.A.; Blackledge, N.P.; Prathapan, P.; Dobrinic, P.; Huseyin, M.K.; et al. Cohesin Disrupts Polycomb-Dependent Chromosome Interactions in Embryonic Stem Cells. Cell Rep. 2020, 30, 820–835.e10. [Google Scholar] [CrossRef] [Green Version]
- Ushiki, A.; Zhang, Y.; Xiong, C.; Zhao, J.; Georgakopoulos-Soares, I.; Kane, L.; Jamieson, K.; Bamshad, M.J.; Nickerson, D.A.; University of Washington Center for Mendelian, G.; et al. Deletion of CTCF sites in the SHH locus alters enhancer-promoter interactions and leads to acheiropodia. Nat. Commun. 2021, 12, 2282. [Google Scholar] [CrossRef]
- Paliou, C.; Guckelberger, P.; Schopflin, R.; Heinrich, V.; Esposito, A.; Chiariello, A.M.; Bianco, S.; Annunziatella, C.; Helmuth, J.; Haas, S.; et al. Preformed chromatin topology assists transcriptional robustness of Shh during limb development. Proc. Natl. Acad. Sci. USA 2019, 116, 12390–12399. [Google Scholar] [CrossRef] [Green Version]
- Gombert, W.M.; Krumm, A. Targeted deletion of multiple CTCF-binding elements in the human C-MYC gene reveals a requirement for CTCF in C-MYC expression. PLoS ONE 2009, 4, e6109. [Google Scholar] [CrossRef]
- de Wit, E.; Vos, E.S.; Holwerda, S.J.; Valdes-Quezada, C.; Verstegen, M.J.; Teunissen, H.; Splinter, E.; Wijchers, P.J.; Krijger, P.H.; de Laat, W. CTCF Binding Polarity Determines Chromatin Looping. Mol. Cell 2015, 60, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Rowley, M.J.; Nichols, M.H.; Lyu, X.; Ando-Kuri, M.; Rivera, I.S.M.; Hermetz, K.; Wang, P.; Ruan, Y.; Corces, V.G. Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 2017, 67, 837–852.e7. [Google Scholar] [CrossRef] [Green Version]
- Matthews, N.E.; White, R. Chromatin Architecture in the Fly: Living withour CTCF/Cohesin Loop Extursion?: Alternating Chromatin States Provide a Basis for Domain Architecture in Drosophila. Bioessays 2019, 41, e1900048. [Google Scholar] [CrossRef] [Green Version]
- Bonchuk, A.; Maksimenko, O.; Kyrchanova, O.; Ivlieva, T.; Mogila, V.; Deshpande, G.; Wolle, D.; Schedl, P.; Georgiev, P. Functional role of dimerization and CP190 interacting domains of CTCF protein in Drosophila melanogaster. BMC Biol. 2015, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Van Bortle, K.; Ramos, E.; Takenaka, N.; Yang, J.; Wahi, J.E.; Corces, V.G. Drosophila CTCF tandemly aligns with other insulator proteins at the borders of H3K27me3 domains. Genome Res. 2012, 22, 2176–2187. [Google Scholar] [CrossRef] [Green Version]
- Cubenas-Potts, C.; Rowley, M.J.; Lyu, X.; Li, G.; Lei, E.P.; Corces, V.G. Different enhancer classes in Drosophila bind distinct architectural proteins and mediate unique chromatin interactions and 3D architecture. Nucleic Acids Res. 2017, 45, 1714–1730. [Google Scholar] [CrossRef]
- Moore, J.M.; Rabaia, N.A.; Smith, L.E.; Fagerlie, S.; Gurley, K.; Loukinov, D.; Disteche, C.M.; Collins, S.J.; Kemp, C.J.; Lobanenkov, V.V.; et al. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS ONE 2012, 7, e34915. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.B.; Pan, H.; Hannenhalli, S.; Cheng, Y.; Ma, J.; Fedoriw, A.; Lobanenkov, V.; Latham, K.E.; Schultz, R.M.; Bartolomei, M.S. Maternal depletion of CTCF reveals multiple functions during oocyte and preimplantation embryo development. Development 2008, 135, 2729–2738. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Tomaszewski, J.M.; Vazquez, M.; Verschoor, S.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- Remeseiro, S.; Cuadrado, A.; Carretero, M.; Martinez, P.; Drosopoulos, W.C.; Canamero, M.; Schildkraut, C.L.; Blasco, M.A.; Losada, A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 2012, 31, 2076–2089. [Google Scholar] [CrossRef]
- Fujita, Y.; Masuda, K.; Bando, M.; Nakato, R.; Katou, Y.; Tanaka, T.; Nakayama, M.; Takao, K.; Miyakawa, T.; Tanaka, T.; et al. Decreased cohesin in the brain leads to defective synapse development and anxiety-related behavior. J. Exp. Med. 2017, 214, 1431–1452. [Google Scholar] [CrossRef] [Green Version]
- De Koninck, M.; Lapi, E.; Badia-Careaga, C.; Cossio, I.; Gimenez-Llorente, D.; Rodriguez-Corsino, M.; Andrada, E.; Hidalgo, A.; Manzanares, M.; Real, F.X.; et al. Essential Roles of Cohesin STAG2 in Mouse Embryonic Development and Adult Tissue Homeostasis. Cell Rep. 2020, 32, 108014. [Google Scholar] [CrossRef]
- Watson, L.A.; Wang, X.; Elbert, A.; Kernohan, K.D.; Galjart, N.; Berube, N.G. Dual effect of CTCF loss on neuroprogenitor differentiation and survival. J. Neurosci. 2014, 34, 2860–2870. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, T.; Tarusawa, E.; Yoshimura, Y.; Galjart, N.; Yagi, T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012, 2, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Sams, D.S.; Nardone, S.; Getselter, D.; Raz, D.; Tal, M.; Rayi, P.R.; Kaphzan, H.; Hakim, O.; Elliott, E. Neuronal CTCF Is Necessary for Basal and Experience-Dependent Gene Regulation, Memory Formation, and Genomic Structure of BDNF and Arc. Cell Rep. 2016, 17, 2418–2430. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Yang, Y.; Valnegri, P.; Juric, I.; Abnousi, A.; Markwalter, K.H.; Guthrie, A.N.; Godec, A.; Oldenborg, A.; Hu, M.; et al. Sensory experience remodels genome architecture in neural circuit to drive motor learning. Nature 2019, 569, 708–713. [Google Scholar] [CrossRef]
- Gomez-Velazquez, M.; Badia-Careaga, C.; Lechuga-Vieco, A.V.; Nieto-Arellano, R.; Tena, J.J.; Rollan, I.; Alvarez, A.; Torroja, C.; Caceres, E.F.; Roy, A.R.; et al. CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLoS Genet. 2017, 13, e1006985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soshnikova, N.; Montavon, T.; Leleu, M.; Galjart, N.; Duboule, D. Functional analysis of CTCF during mammalian limb development. Dev. Cell 2010, 19, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, H.; Ribeiro de Almeida, C.; Sleutels, F.; Dingjan, G.; van de Nobelen, S.; Jonkers, I.; Ling, K.W.; Gribnau, J.; Renkawitz, R.; Grosveld, F.; et al. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 2008, 27, 2839–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.; Kim, H.P.; Shin, J.O. Depletion of CTCF induces craniofacial malformations in mouse embryos. Am. J. Transl. Res. 2019, 11, 6102–6109. [Google Scholar] [PubMed]
- Seitan, V.C.; Hao, B.; Tachibana-Konwalski, K.; Lavagnolli, T.; Mira-Bontenbal, H.; Brown, K.E.; Teng, G.; Carroll, T.; Terry, A.; Horan, K.; et al. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 2011, 476, 467–471. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.B.; Peterson, J.; Reimer, M.; Stelloh, C.; Pulakanti, K.; Gerbec, Z.J.; Abel, A.M.; Strouse, J.M.; Strouse, C.; McNulty, M.; et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 2017, 31, 712–719. [Google Scholar] [CrossRef]
- Gregor, A.; Oti, M.; Kouwenhoven, E.N.; Hoyer, J.; Sticht, H.; Ekici, A.B.; Kjaergaard, S.; Rauch, A.; Stunnenberg, H.G.; Uebe, S.; et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am. J. Hum. Genet. 2013, 93, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Bastaki, F.; Nair, P.; Mohamed, M.; Malik, E.M.; Helmi, M.; Al-Ali, M.T.; Hamzeh, A.R. Identification of a novel CTCF mutation responsible for syndromic intellectual disability—A case report. BMC Med. Genet. 2017, 18, 68. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Yuan, H.; Wu, W.; Chen, S.; Yang, Q.; Wang, J.; Zhang, Q.; Gui, B.; Fan, X.; Chen, R.; et al. Three additional de novo CTCF mutations in Chinese patients help to define an emerging neurodevelopmental disorder. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 218–225. [Google Scholar] [CrossRef]
- Hori, I.; Kawamura, R.; Nakabayashi, K.; Watanabe, H.; Higashimoto, K.; Tomikawa, J.; Ieda, D.; Ohashi, K.; Negishi, Y.; Hattori, A.; et al. CTCF deletion syndrome: Clinical features and epigenetic delineation. J. Med. Genet. 2017, 54, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hoekzema, K.; Vecchio, D.; Wu, H.; Sulovari, A.; Coe, B.P.; Gillentine, M.A.; Wilfert, A.B.; Perez-Jurado, L.A.; Kvarnung, M.; et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 2020, 11, 4932. [Google Scholar] [CrossRef] [PubMed]
- Konrad, E.D.H.; Nardini, N.; Caliebe, A.; Nagel, I.; Young, D.; Horvath, G.; Santoro, S.L.; Shuss, C.; Ziegler, A.; Bonneau, D.; et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 2019, 21, 2723–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraide, T.; Yamoto, K.; Masunaga, Y.; Asahina, M.; Endoh, Y.; Ohkubo, Y.; Matsubayashi, T.; Tsurui, S.; Yamada, H.; Yanagi, K.; et al. Genetic and phenotypic analysis of 101 patients with developmental delay or intellectual disability using whole-exome sequencing. Clin. Genet. 2021, 100, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Brunet, T.; Jech, R.; Brugger, M.; Kovacs, R.; Alhaddad, B.; Leszinski, G.; Riedhammer, K.M.; Westphal, D.S.; Mahle, I.; Mayerhanser, K.; et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin. Genet. 2021, 100, 14–28. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders, S. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef]
- Meng, L.; Pammi, M.; Saronwala, A.; Magoulas, P.; Ghazi, A.R.; Vetrini, F.; Zhang, J.; He, W.; Dharmadhikari, A.V.; Qu, C.; et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr. 2017, 171, e173438. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Geisheker, M.R.; Heymann, G.; Wang, T.; Coe, B.P.; Turner, T.N.; Stessman, H.A.F.; Hoekzema, K.; Kvarnung, M.; Shaw, M.; Friend, K.; et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat. Neurosci. 2017, 20, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Cappi, C.; Oliphant, M.E.; Peter, Z.; Zai, G.; Conceicao do Rosario, M.; Sullivan, C.A.W.; Gupta, A.R.; Hoffman, E.J.; Virdee, M.; Olfson, E.; et al. De Novo Damaging DNA Coding Mutations Are Associated With Obsessive-Compulsive Disorder and Overlap With Tourette’s Disorder and Autism. Biol. Psychiatry 2020, 87, 1035–1044. [Google Scholar] [CrossRef]
- Yuen, R.K.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 2016, 1, 160271–1602710. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Monnich, M.; Yan, Y.; Xu, W.; Gil-Rodriguez, M.C.; et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minor, A.; Shinawi, M.; Hogue, J.S.; Vineyard, M.; Hamlin, D.R.; Tan, C.; Donato, K.; Wysinger, L.; Botes, S.; Das, S.; et al. Two novel RAD21 mutations in patients with mild Cornelia de Lange syndrome-like presentation and report of the first familial case. Gene 2014, 537, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Pereza, N.; Severinski, S.; Ostojic, S.; Volk, M.; Maver, A.; Dekanic, K.B.; Kapovic, M.; Peterlin, B. Cornelia de Lange syndrome caused by heterozygous deletions of chromosome 8q24: Comments on the article by Pereza et al. [2012]. Am. J. Med. Genet. A 2015, 167, 1426–1427. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Poke, G.; Ferry, Q.; Williamson, K.; Aldridge, R.; Meynert, A.M.; Bengani, H.; Chan, C.Y.; Kayserili, H.; Avci, S.; et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet. 2014, 51, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.I.; Jespersgaard, C.; Nazaryan, L.; Bisgaard, A.M.; Tumer, Z. A novel RAD21 variant associated with intrafamilial phenotypic variation in Cornelia de Lange syndrome-review of the literature. Clin. Genet. 2017, 91, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Anneren, G.; Marcos-Alcalde, I.; Wilbe, M.; Melin, M.; Gomez-Puertas, P.; Bondeson, M.L. A novel RAD21 p.(Gln592del) variant expands the clinical description of Cornelia de Lange syndrome type 4-Review of the literature. Eur. J. Med. Genet. 2019, 62, 103526. [Google Scholar] [CrossRef]
- Dorval, S.; Masciadri, M.; Mathot, M.; Russo, S.; Revencu, N.; Larizza, L. A novel RAD21 mutation in a boy with mild Cornelia de Lange presentation: Further delineation of the phenotype. Eur. J. Med. Genet. 2020, 63, 103620. [Google Scholar] [CrossRef]
- Goel, H.; Parasivam, G. Another case of holoprosencephaly associated with RAD21 loss-of-function variant. Brain 2020, 143, e64. [Google Scholar] [CrossRef]
- Bonora, E.; Bianco, F.; Cordeddu, L.; Bamshad, M.; Francescatto, L.; Dowless, D.; Stanghellini, V.; Cogliandro, R.F.; Lindberg, G.; Mungan, Z.; et al. Mutations in RAD21 disrupt regulation of APOB in patients with chronic intestinal pseudo-obstruction. Gastroenterology 2015, 148, 771–782.e11. [Google Scholar] [CrossRef]
- Lee, C.J.; Evans, J.; Kim, K.; Chae, H.; Kim, S. Determining the effect of DNA methylation on gene expression in cancer cells. In Gene Function Analysis; Humana Press: Totowa, NJ, USA, 2014; Volume 1101, pp. 161–178. [Google Scholar] [CrossRef]
- Martinez, F.; Caro-Llopis, A.; Rosello, M.; Oltra, S.; Mayo, S.; Monfort, S.; Orellana, C. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J. Med. Genet. 2017, 54, 87–92. [Google Scholar] [CrossRef]
- McBrien, J.; Crolla, J.A.; Huang, S.; Kelleher, J.; Gleeson, J.; Lynch, S.A. Further case of microdeletion of 8q24 with phenotype overlapping Langer-Giedion without TRPS1 deletion. Am. J. Med. Genet. A 2008, 146A, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.E.; Patel, V.; Jin, W.; Adam, M.P.; et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. 2019, 21, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Krab, L.C.; Marcos-Alcalde, I.; Assaf, M.; Balasubramanian, M.; Andersen, J.B.; Bisgaard, A.M.; Fitzpatrick, D.R.; Gudmundsson, S.; Huisman, S.A.; Kalayci, T.; et al. Delineation of phenotypes and genotypes related to cohesin structural protein RAD21. Hum. Genet. 2020, 139, 575–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musio, A.; Selicorni, A.; Focarelli, M.L.; Gervasini, C.; Milani, D.; Russo, S.; Vezzoni, P.; Larizza, L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006, 38, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodriguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Gil-Rodriguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernandez-Marcos, M.; Teresa-Rodrigo, M.E.; et al. De novo heterozygous mutations in SMC3 cause a range of Cornelia de Lange syndrome-overlapping phenotypes. Hum. Mutat. 2015, 36, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Hoppman-Chaney, N.; Jang, J.S.; Jen, J.; Babovic-Vuksanovic, D.; Hodge, J.C. In-frame multi-exon deletion of SMC1A in a severely affected female with Cornelia de Lange Syndrome. Am. J. Med. Genet. A 2012, 158A, 193–198. [Google Scholar] [CrossRef]
- Limongelli, G.; Russo, S.; Digilio, M.C.; Masciadri, M.; Pacileo, G.; Fratta, F.; Martone, F.; Maddaloni, V.; D’Alessandro, R.; Calabro, P.; et al. Hypertrophic cardiomyopathy in a girl with Cornelia de Lange syndrome due to mutation in SMC1A. Am. J. Med. Genet. A 2010, 152A, 2127–2129. [Google Scholar] [CrossRef]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [Green Version]
- Wenger, T.L.; Chow, P.; Randle, S.C.; Rosen, A.; Birgfeld, C.; Wrede, J.; Javid, P.; King, D.; Manh, V.; Hing, A.V.; et al. Novel findings of left ventricular non-compaction cardiomyopathy, microform cleft lip and poor vision in patient with SMC1A-associated Cornelia de Lange syndrome. Am. J. Med. Genet. A 2017, 173, 414–420. [Google Scholar] [CrossRef]
- Pie, J.; Gil-Rodriguez, M.C.; Ciero, M.; Lopez-Vinas, E.; Ribate, M.P.; Arnedo, M.; Deardorff, M.A.; Puisac, B.; Legarreta, J.; de Karam, J.C.; et al. Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome. Am. J. Med. Genet. A 2010, 152A, 924–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervasini, C.; Russo, S.; Cereda, A.; Parenti, I.; Masciadri, M.; Azzollini, J.; Melis, D.; Aravena, T.; Doray, B.; Ferrarini, A.; et al. Cornelia de Lange individuals with new and recurrent SMC1A mutations enhance delineation of mutation repertoire and phenotypic spectrum. Am. J. Med. Genet. A 2013, 161A, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Kleefstra, T.; Willemsen, M.H.; de Vries, P.; Pfundt, R.; Hehir-Kwa, J.Y.; Gilissen, C.; Veltman, J.A.; de Vries, B.B.; Vissers, L.E. De novo loss-of-function mutations in X-linked SMC1A cause severe ID and therapy-resistant epilepsy in females: Expanding the phenotypic spectrum. Clin. Genet. 2016, 90, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feldman, R.; Zhang, Z.; Deardorff, M.A.; Haverfield, E.V.; Kaur, M.; Li, J.R.; Clark, D.; Kline, A.D.; Waggoner, D.J.; et al. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum. Mutat. 2009, 30, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Borck, G.; Zarhrate, M.; Bonnefont, J.P.; Munnich, A.; Cormier-Daire, V.; Colleaux, L. Incidence and clinical features of X-linked Cornelia de Lange syndrome due to SMC1L1 mutations. Hum. Mutat. 2007, 28, 205–206. [Google Scholar] [CrossRef]
- Lehalle, D.; Mosca-Boidron, A.L.; Begtrup, A.; Boute-Benejean, O.; Charles, P.; Cho, M.T.; Clarkson, A.; Devinsky, O.; Duffourd, Y.; Duplomb-Jego, L.; et al. STAG1 mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J. Med. Genet. 2017, 54, 479–488. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Mulatinho, M.V.; Senaratne, T.N.; Singh, K.; Center, U.C.G.; Nguyen, D.C.; Gallant, N.M.; Strom, S.P.; Ghahremani, S.; et al. De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies. Am. J. Med. Genet. A 2017, 173, 1319–1327. [Google Scholar] [CrossRef]
- Soardi, F.C.; Machado-Silva, A.; Linhares, N.D.; Zheng, G.; Qu, Q.; Pena, H.B.; Martins, T.M.M.; Vieira, H.G.S.; Pereira, N.B.; Melo-Minardi, R.C.; et al. Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ Genom. Med. 2017, 2, 7. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Signer, R.H.; Center, U.C.G.; Vilain, E.; Martinez-Agosto, J.A. Mutations in STAG2 cause an X-linked cohesinopathy associated with undergrowth, developmental delay, and dysmorphia: Expanding the phenotype in males. Mol. Genet. Genom. Med. 2019, 7, e00501. [Google Scholar] [CrossRef] [Green Version]
- Epilepsy Genetics, I. The Epilepsy Genetics Initiative: Systematic reanalysis of diagnostic exomes increases yield. Epilepsia 2019, 60, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Freyberger, F.; Kokotovic, T.; Krnjak, G.; Frkovic, S.H.; Nagy, V. Expanding the known phenotype of Mullegama-Klein-Martinez syndrome in male patients. Hum. Genome Var. 2021, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Sawle, A.D.; Wynn, J.; Aspelund, G.; Stolar, C.J.; Arkovitz, M.S.; Potoka, D.; Azarow, K.S.; Mychaliska, G.B.; Shen, Y.; et al. Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum. Mol. Genet. 2015, 24, 4764–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kruszka, P.; Berger, S.I.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Prijoles, E.J.; et al. Cohesin complex-associated holoprosencephaly. Brain 2019, 142, 2631–2643. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. Cancer 2020, 20, 504–515. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Carico, Z.M.; Stefan, H.C.; Justice, M.; Yimit, A.; Dowen, J.M. A cohesin cancer mutation reveals a role for the hinge domain in genome organization and gene expression. PLoS Genet. 2021, 17, e1009435. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cummings, C.T.; Rowley, M.J. Implications of Dosage Deficiencies in CTCF and Cohesin on Genome Organization, Gene Expression, and Human Neurodevelopment. Genes 2022, 13, 583. https://doi.org/10.3390/genes13040583
Cummings CT, Rowley MJ. Implications of Dosage Deficiencies in CTCF and Cohesin on Genome Organization, Gene Expression, and Human Neurodevelopment. Genes. 2022; 13(4):583. https://doi.org/10.3390/genes13040583
Chicago/Turabian StyleCummings, Christopher T., and M. Jordan Rowley. 2022. "Implications of Dosage Deficiencies in CTCF and Cohesin on Genome Organization, Gene Expression, and Human Neurodevelopment" Genes 13, no. 4: 583. https://doi.org/10.3390/genes13040583