
Expanding the Phenotype of B3GALNT2-Related Disorders


 and
and
Abstract
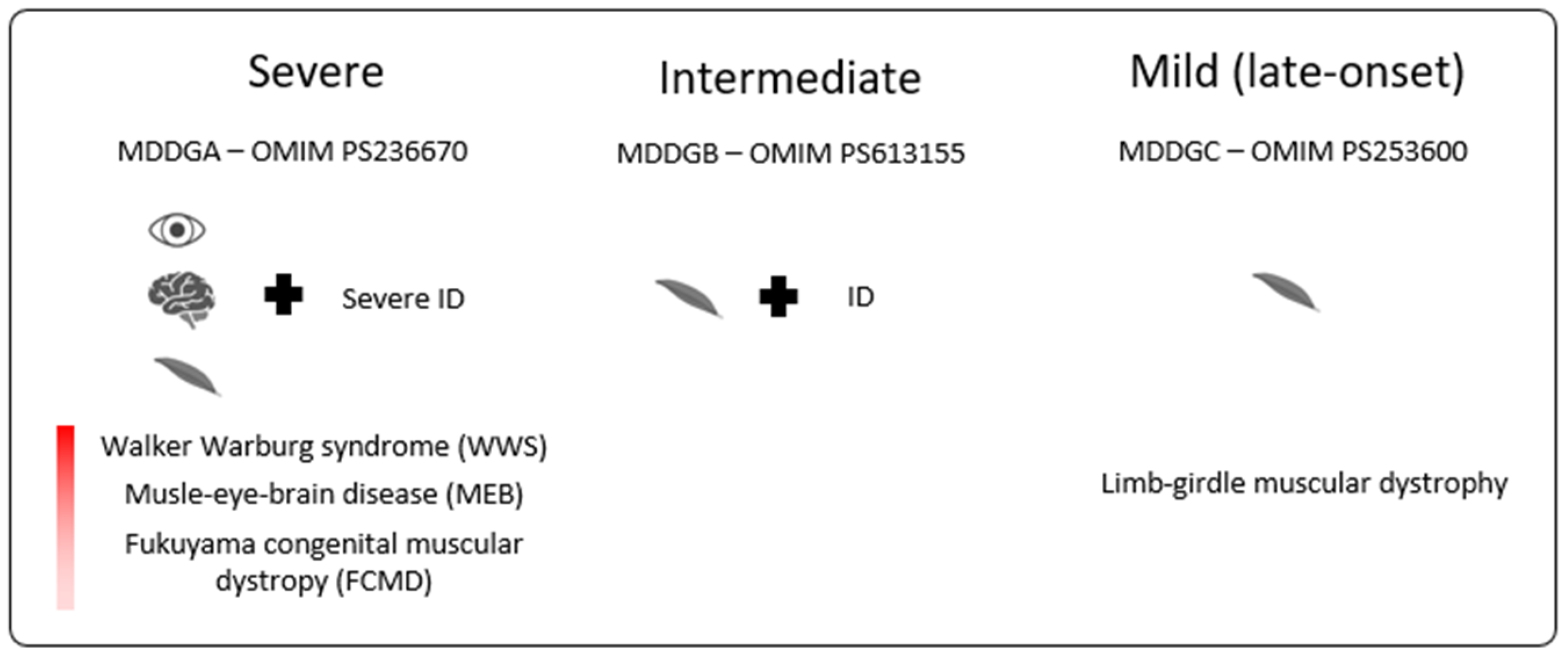
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Exome Sequencing (ES)
2.3. Array-CGH
2.4. SNP-Array
3. Results
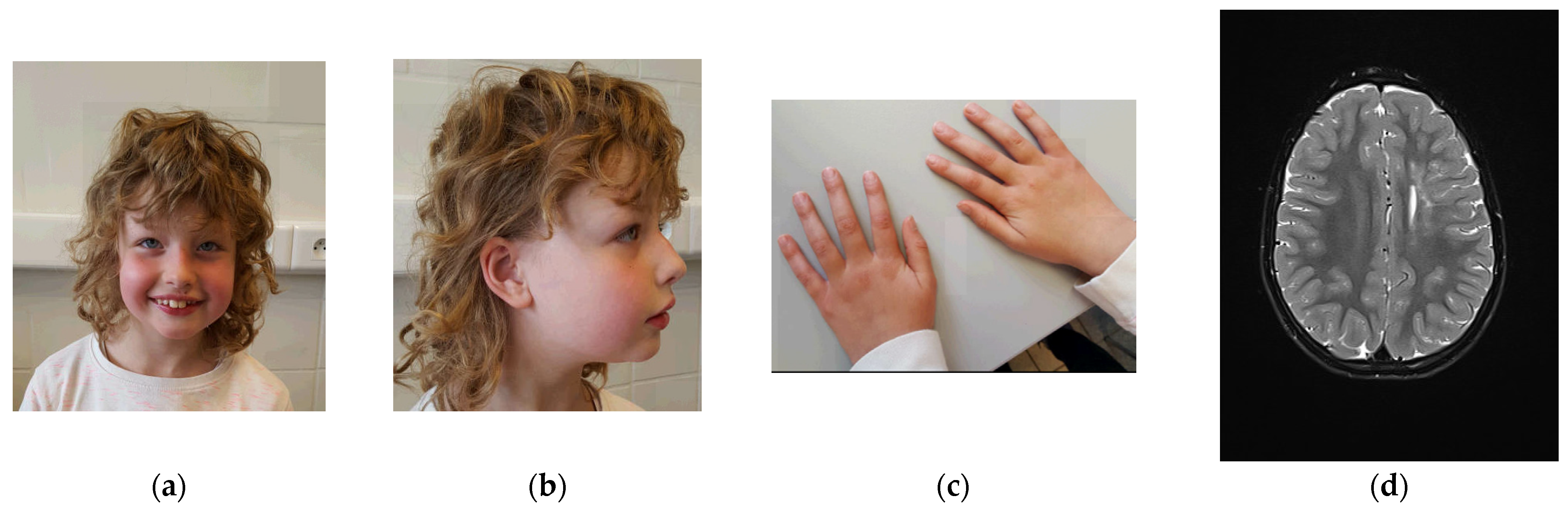
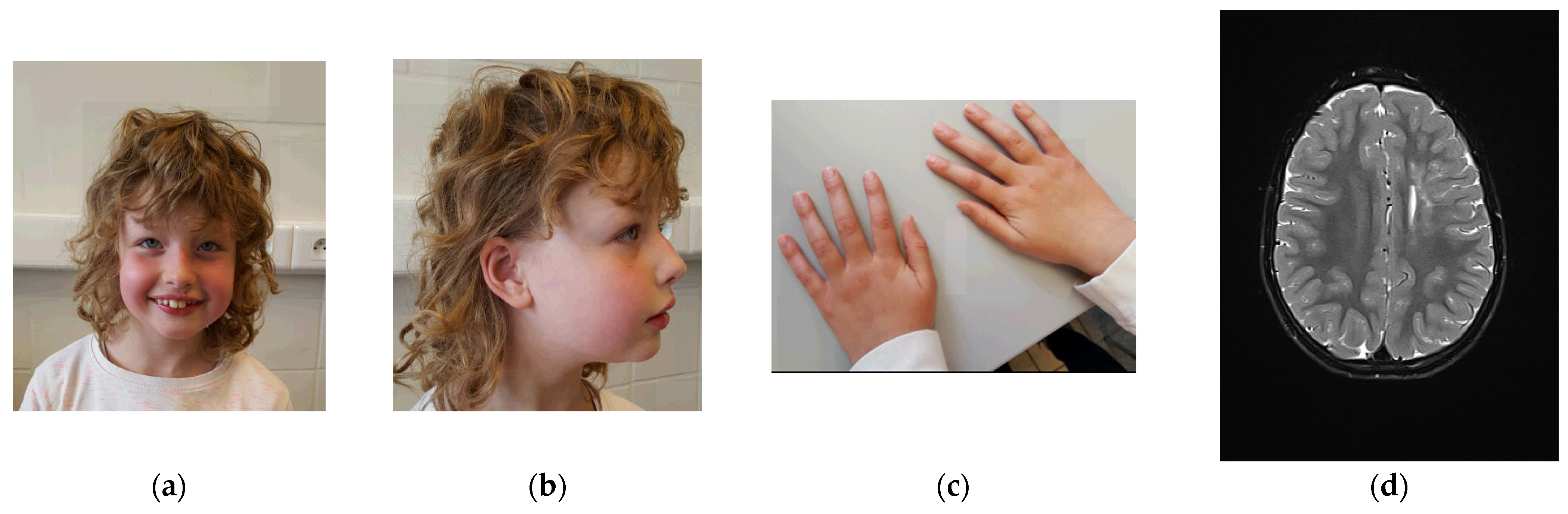
3.1. Clinical Description
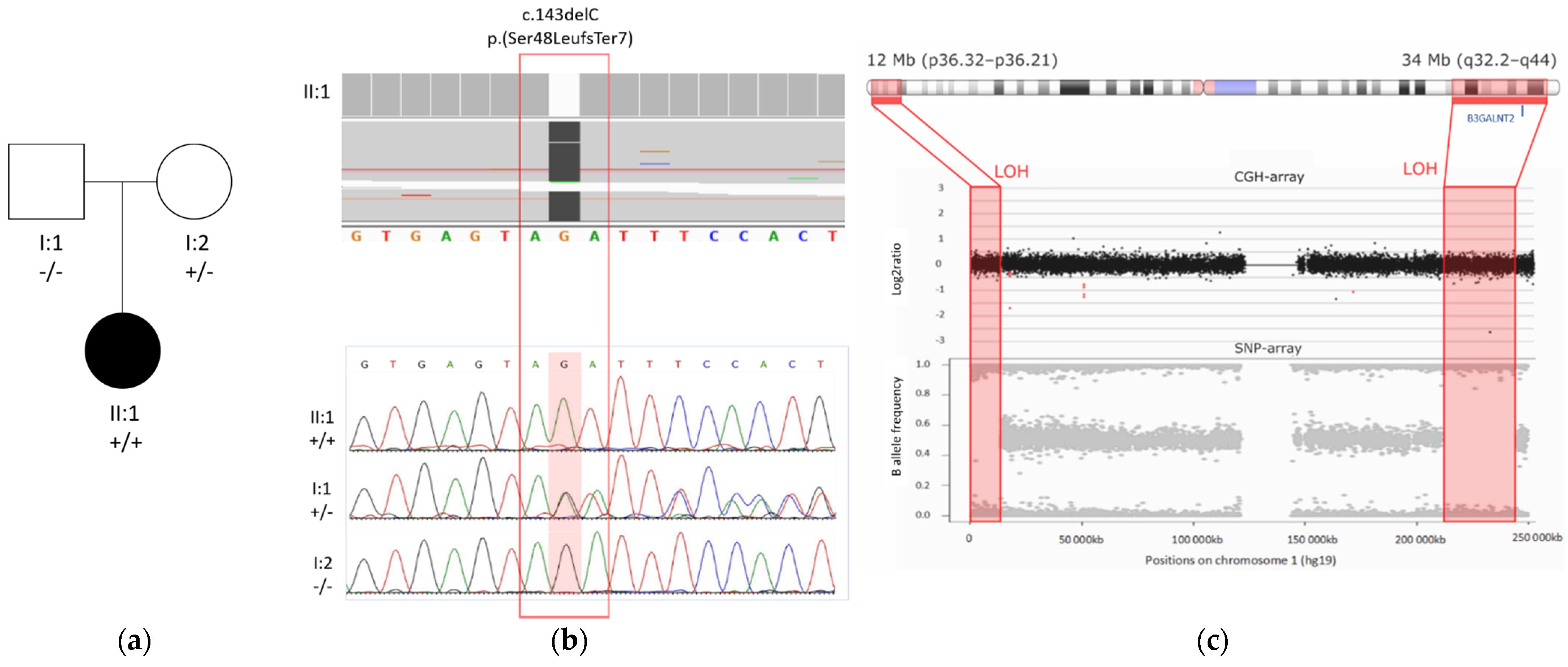
3.2. Molecular Result
3.3. Review of Previously Reported Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pilgram, G.S.; Potikanond, S.; Baines, R.A.; Fradkin, L.G.; Noordermeer, J.N. The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol. Neurobiol. 2010, 41, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, E.; Mercuri, E.; Godfrey, C.; Smith, J.; Robb, S.; Kinali, M.; Straub, V.; Bushby, K.; Manzur, A.; Talim, B.; et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol. 2008, 64, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.E. Abnormal glycosylation of dystroglycan in human genetic disease. Biochim. Biophys. Acta 2009, 1792, 853–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.T. Dystroglycan glycosylation and its role in matrix binding in skeletal muscle. Glycobiology 2003, 13, 55R–66R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchet-Seraphin, C.; Vuillaumier-Barrot, S.; Seta, N. Dystroglycanopathies: About Numerous Genes Involved in Glycosylation of One Single Glycoprotein. J. Neuromuscul. Dis. 2015, 2, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagawa, M.; Toda, T. Muscular Dystrophy with Ribitol-Phosphate Deficiency: A Novel Post-Translational Mechanism in Dystroglycanopathy. J. Neuromuscul. Dis. 2017, 4, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida-Moriguchi, T.; Willer, T.; Anderson, M.E.; Venzke, D.; Whyte, T.; Muntoni, F.; Lee, H.; Nelson, S.F.; Yu, L.; Campbell, K.P. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 2013, 341, 896–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, E.; Carss, K.J.; Cirak, S.; Foley, A.R.; Torelli, S.; Willer, T.; Tambunan, D.E.; Yau, S.; Brodd, L.; Sewry, C.A.; et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am. J. Hum. Genet. 2013, 92, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.L.; Glenn, O.A.; Sherr, E.H.; Strober, J.B. Serial prenatal and postnatal MRI of dystroglycanopathy in a patient with familial B3GALNT2 mutation. Pediatric Radiol. 2017, 47, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Maroofian, R.; Riemersma, M.; Jae, L.T.; Zhianabed, N.; Willemsen, M.H.; Wissink-Lindhout, W.M.; Willemsen, M.A.; de Brouwer, A.P.M.; Mehrjardi, M.Y.V.; Ashrafi, M.R.; et al. B3GALNT2 mutations associated with non-syndromic autosomal recessive intellectual disability reveal a lack of genotype-phenotype associations in the muscular dystrophy-dystroglycanopathies. Genome Med. 2017, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Dhaibani, M.A.; El-Hattab, A.W.; Ismayl, O.; Suleiman, J. B3GALNT2-Related Dystroglycanopathy: Expansion of the Phenotype with Novel Mutation Associated with Muscle-Eye-Brain Disease, Walker-Warburg Syndrome, Epileptic Encephalopathy-West Syndrome, and Sensorineural Hearing Loss. Neuropediatrics 2018, 49, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, C.; Oldfors, A.; Darin, N. B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations. Eur. J. Hum. Genet. EJHG 2014, 22, 707–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauy, K.; de Leeuw, N.; Yntema, H.G.; Pfundt, R.; Gilissen, C. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buysse, K.; Delle Chiaie, B.; Van Coster, R.; Loeys, B.; De Paepe, A.; Mortier, G.; Speleman, F.; Menten, B. Challenges for CNV interpretation in clinical molecular karyotyping: Lessons learned from a 1001 sample experience. Eur. J. Med. Genet. 2009, 52, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: Consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 2011, 31, 595–613. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
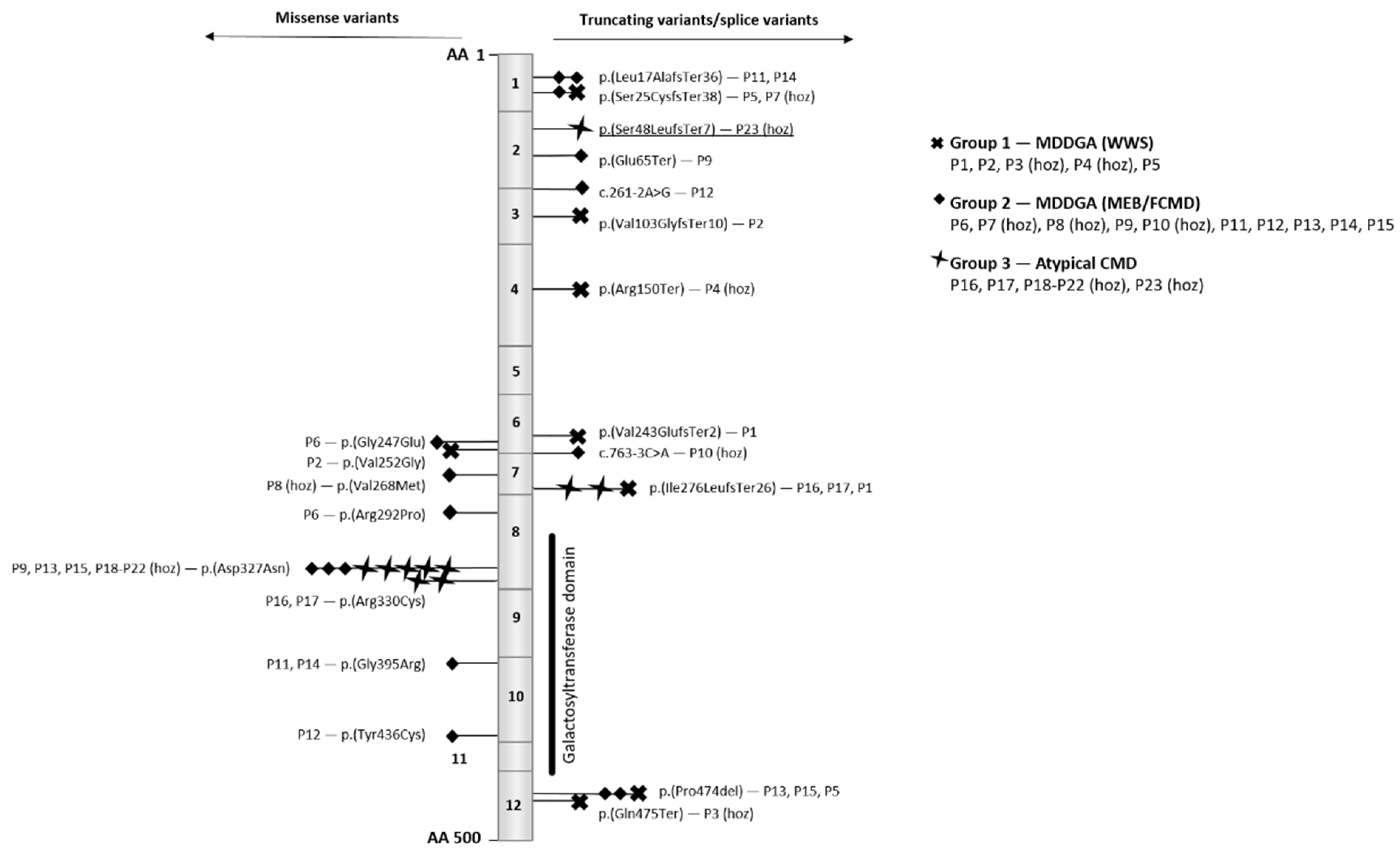{kind=link}
| Patient Number | c. and p. Position of Variant | Type Variant | Phenotype |
|---|---|---|---|
| P1 | c.726_727del; p.(Val243GlufsTer2) c.822_823dup; p.(Ile276LeufsTer26) | Frameshift Frameshift | WWS (MDDGA) |
| P2 | c.308_309del; p.(Val103GlyfsTer10) c.755T > G; p.(Val252Gly) | Frameshift Missense | WWS (MDDGA) |
| P3 | c.1423C > T; p.(Gln475Ter) | Nonsense (hoz) | WWS (MDDGA) |
| P4 | c.448C > T; p.(Arg150Ter) | Nonsense (hoz) | WWS (MDDGA) |
| P5 | c.51_73dup; p.(Ser25CysfsTer38) c.1421_1423delCTC; p.(Pro474del) | Frameshift In-frame | WWS (MDDGA) |
| P6 | c.740G > C; p.(Gly247Glu) c.875G > C; p.(Arg292Pro) | Missense Missense | MEB/FCMD (MDDGA) |
| P7 | c.51_73dup; p.(Ser25CysfsTer38) | Frameshift (hoz) | MEB/FCMD (MDDGA) |
| P8 | c.802G > A; p.(Val268Met) | Missense (hoz) | MEB/FCMD (MDDGA) |
| P9 | c.192dupT; p.(Glu65Ter) c.979G > A; p.(Asp327Asn) | Nonsense Missense | MEB/FCMD (MDDGA) |
| P10 | c.763-3C > A | Splice acceptor (hoz) | MEB/FCMD (MDDGA) |
| P11 | c.48dup; p.(Leu17AlafsTer36) c.1183G > A; p.(Gly395Arg) | Frameshift Missense | MEB/FCMD (MDDGA) |
| P12 | c.261-2A > G c.1307A > G; p.(Tyr436Cys) | Splice donor Missense | MEB/FCMD (MDDGA) |
| P13 | c.979G > A; p.(Asp327Asn) c.1421_1423delCTC; p.(Pro474del) | Missense In-frame | MEB/FCMD (MDDGA) |
| P14 | c.48dupG; p.(Leu17fsTer36) c.1183G > A; p.(Gly395Arg) | Frameshift Missense | MEB/FCMD (MDDGA) |
| P15 | c.979G > A; p.(Asp327Asn) c.1421_1423delCTC; p.(Pro474del) | Missense In-frame | MEB/FCMD (MDDGA) |
| P16 | c.822_823dup; p.(Ile276LeufsTer26) c.988C > T; p.(Arg330Cys) | Frameshift Missense | Atypical CMD |
| P17 | c.822_823dup; p.(Ile276LeufsTer26) c.988C > T; p.(Arg330Cys) | Frameshift Missense | Atypical CMD |
| P18 | c.979G > A; p.(Asp327Asn) | Missense (hoz) | Atypical CMD |
| P19 | c.979G > A; p.(Asp327Asn) | Missense (hoz) | Atypical CMD |
| P20 | c.979G > A; p.(Asp327Asn) | Missense (hoz) | Atypical CMD |
| P21 | c.979G > A; p.(Asp327Asn) | Missense (hoz) | Atypical CMD |
| P22 | c.979G > A; p.(Asp327Asn) | Missense (hoz) | Atypical CMD |
| P23 (our case) | c.143del; p.(Ser48LeufsTer7) | Frameshift (hoz) | Atypical CMD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’haenens, E.; Vergult, S.; Menten, B.; Dheedene, A.; Kooy, R.F.; Callewaert, B. Expanding the Phenotype of B3GALNT2-Related Disorders. Genes 2022, 13, 694. https://doi.org/10.3390/genes13040694
D’haenens E, Vergult S, Menten B, Dheedene A, Kooy RF, Callewaert B. Expanding the Phenotype of B3GALNT2-Related Disorders. Genes. 2022; 13(4):694. https://doi.org/10.3390/genes13040694
Chicago/Turabian StyleD’haenens, Erika, Sarah Vergult, Björn Menten, Annelies Dheedene, R. Frank Kooy, and Bert Callewaert. 2022. "Expanding the Phenotype of B3GALNT2-Related Disorders" Genes 13, no. 4: 694. https://doi.org/10.3390/genes13040694
APA StyleD’haenens, E., Vergult, S., Menten, B., Dheedene, A., Kooy, R. F., & Callewaert, B. (2022). Expanding the Phenotype of B3GALNT2-Related Disorders. Genes, 13(4), 694. https://doi.org/10.3390/genes13040694