
Specific Gain and Loss of Co-Expression Modules in Long-Lived Individuals Indicate a Role of circRNAs in Human Longevity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and RNA-seq
2.2. CircRNA Prediction and Quantification
2.3. Analysis of Cell-Type Composition and General circRNA Expression Pattern
2.4. Construction of circRNA-circRNA Co-Expression Network and Module Preservation Analysis
2.5. CircRNA-Gene (mRNA) Co-Expression Analysis and Gene Enrichment Analysis
3. Results
3.1. Identification and General Expression Pattern of circRNAs in Longevous Families
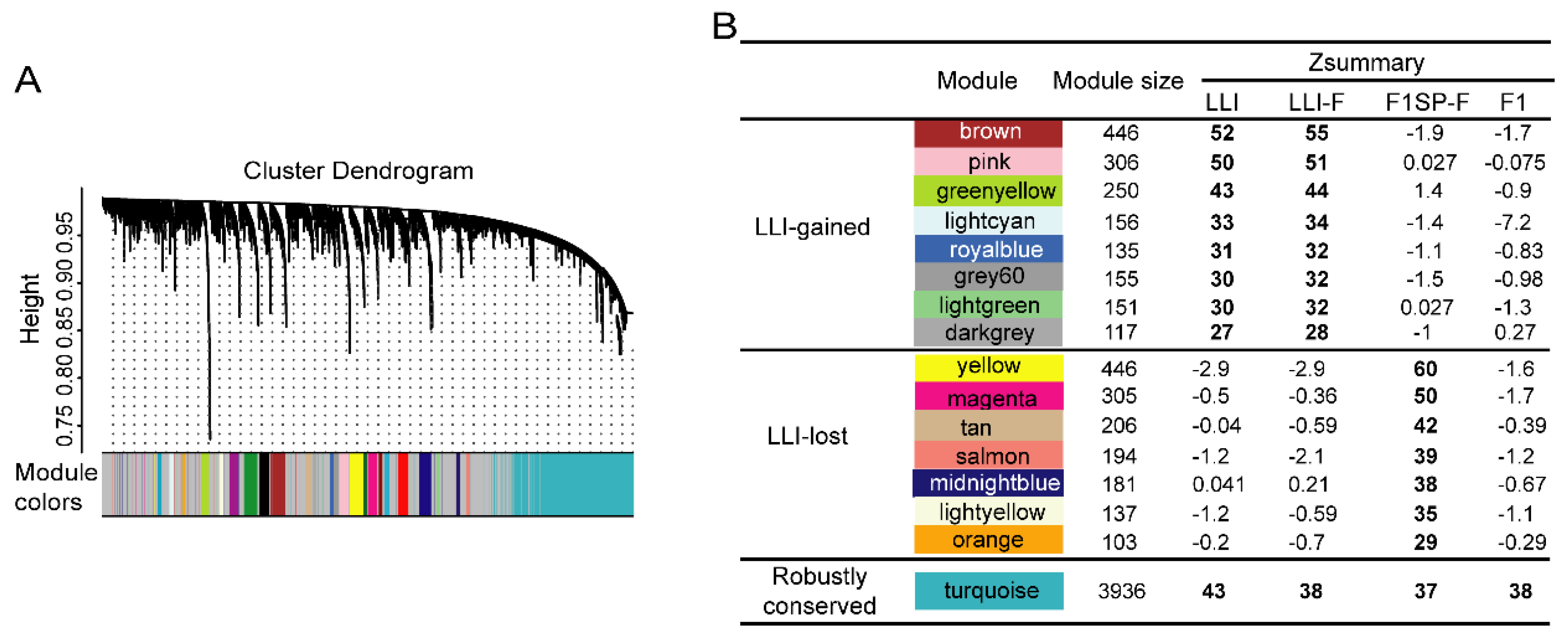
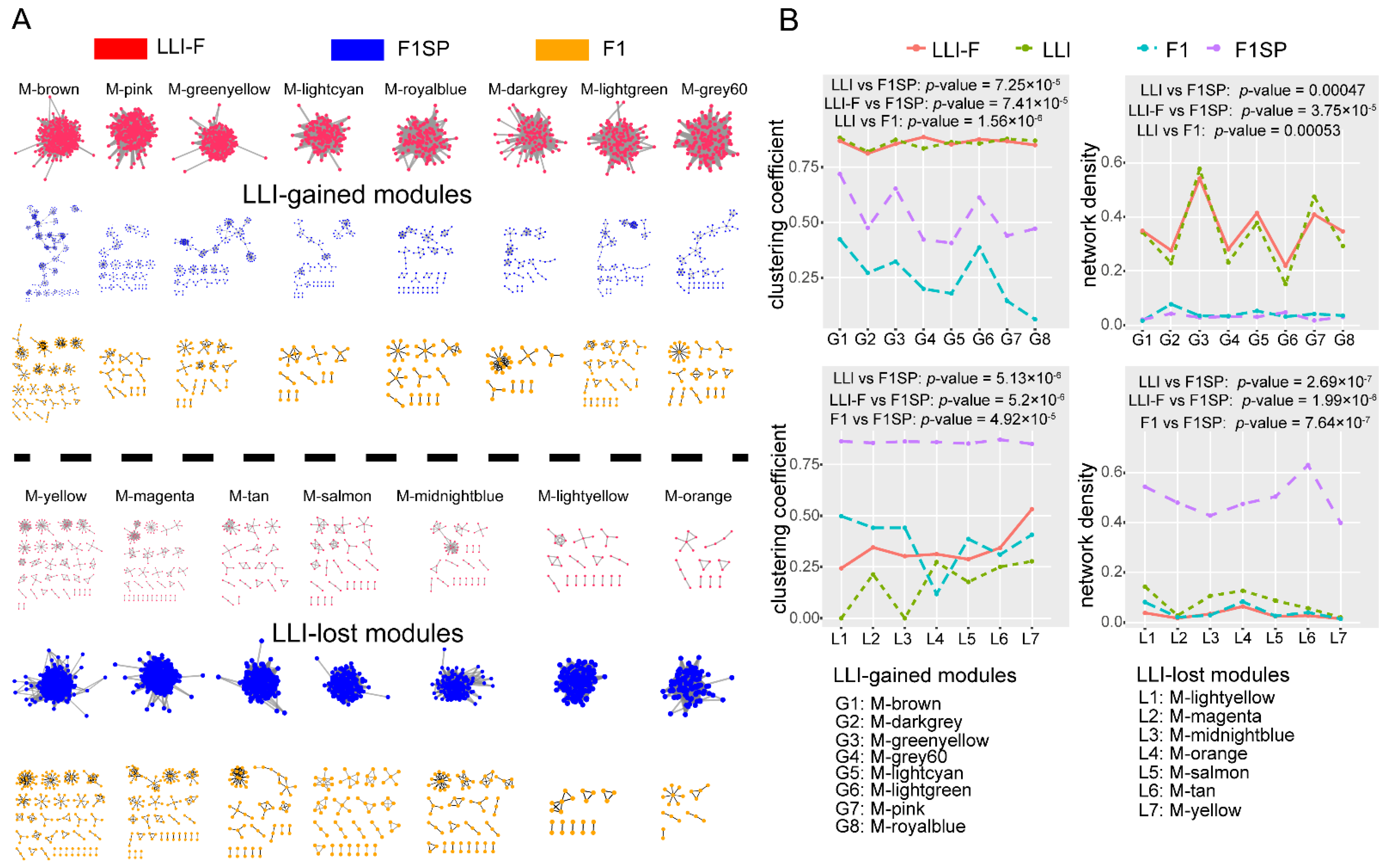
3.2. Specific circRNAs Co-Expression Modules Were Observed Gained or Lost in Long-Lived Individuals
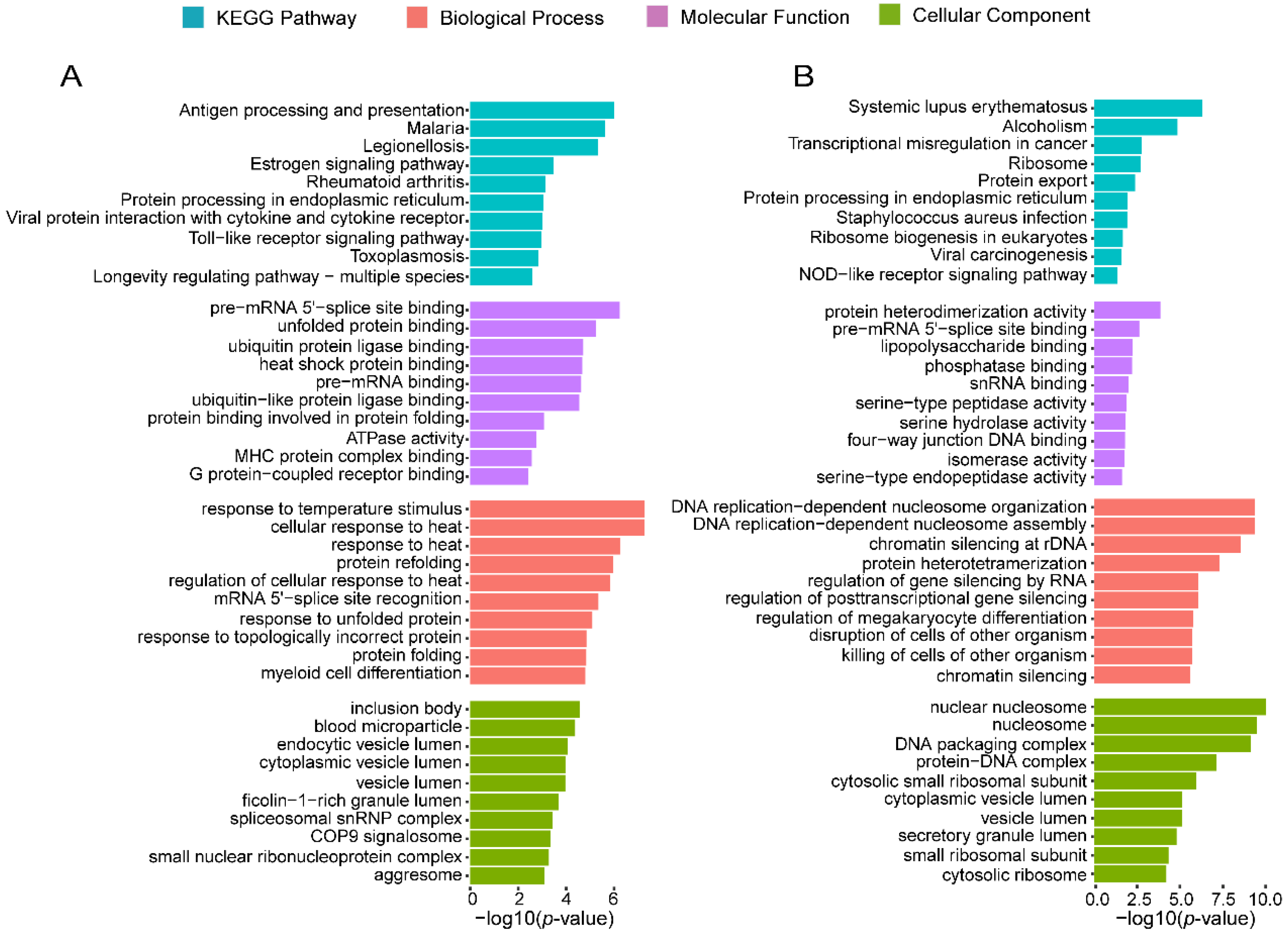
3.3. LLI-Gained/Lost c-CCMs Were Proved to Be Related to Healthy Aging-Related Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, Y.; Xiong, W.; Zhang, Z.; Wang, Z.; Lv, L.; Liu, C.; Hu, Z.; Zheng, Y.T.; Lu, L.; et al. CircGRIA1 shows an age-related increase in male macaque brain and regulates synaptic plasticity and synaptogenesis. Nat. Commun. 2020, 11, 3594. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.H.; Chen, X.Q.; Yu, Q.; Ye, Y.; Liu, Y.W.; Yan, D.; Yang, L.Q.; Chen, G.; Lin, R.; Yang, L.; et al. Transcriptome evidence reveals enhanced autophagy-lysosomal function in centenarians. Genome Res. 2018, 28, 1601–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, F.H.; Chen, X.Q.; He, Y.H.; Kong, Q.P. Accelerated DNA methylation changes in middle-aged men define sexual dimorphism in human lifespans. Clin. Epigenetics 2018, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Li, G.H.; Han, F.; Xiao, F.H.; Gu, K.S.; Shen, Q.; Xu, W.; Li, W.X.; Wang, Y.L.; Liang, B.; Huang, J.F.; et al. System-level metabolic modeling facilitates unveiling metabolic signature in exceptional longevity. Aging Cell 2022, 21, e13595. [Google Scholar] [CrossRef]
- Jiang, J.; Cheng, L.; Yan, L.; Ge, M.; Yang, L.; Ying, H.; Kong, Q. Decoding the role of long noncoding RNAs in the healthy aging of centenarians. Brief. Bioinform. 2021, 22, bbaa439. [Google Scholar] [CrossRef]
- Jiang, J.J.; Cheng, L.H.; Wu, H.; He, Y.H.; Kong, Q.P. Insights into long noncoding RNAs of naked mole rat (Heterocephalus glaber) and their potential association with cancer resistance. Epigenetics Chromatin 2016, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.J.; Kong, Q.P. Comparative analysis of long noncoding RNAs in long-lived mammals provides insights into natural cancer-resistance. RNA Biol. 2020, 17, 1657–1665. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Zhang, C.; Yang, W.; Yong, T.; Awan, F.M.; Yang, B.B. Identifying and Characterizing circRNA-Protein Interaction. Theranostics 2017, 7, 4183–4191. [Google Scholar] [CrossRef] [PubMed]
- Patop, I.L.; Wust, S.; Kadener, S. Past, present, and future of circRNAs. EMBO J. 2019, 38, e100836. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell. Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Dube, U.; Del-Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; Norton, J.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef]
- Garikipati, V.N.S.; Verma, S.K.; Cheng, Z.; Liang, D.; Truongcao, M.M.; Cimini, M.; Yue, Y.; Huang, G.; Wang, C.; Benedict, C.; et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 2019, 10, 4317. [Google Scholar] [CrossRef]
- Stoll, L.; Rodríguez-Trejo, A.; Guay, C.; Brozzi, F.; Bayazit, M.B.; Gattesco, S.; Menoud, V.; Sobel, J.; Marques, A.C.; Venø, M.T.; et al. A circular RNA generated from an intron of the insulin gene controls insulin secretion. Nat. Commun. 2020, 11, 5611. [Google Scholar] [CrossRef]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e13. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, X.; Zhang, M.; Yan, S.; Sun, C.; Xiao, F.; Huang, N.; Yang, X.; Zhao, K.; Zhou, H.; et al. Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. JNCI J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.; Ma, X.K.; Li, G.W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genom. Proteom. Bioinform. 2018, 16, 226–233. [Google Scholar] [CrossRef]
- MacInnes, A.W. The role of the ribosome in the regulation of longevity and lifespan extension. Wiley Interdiscip Rev. RNA 2016, 7, 198–212. [Google Scholar] [CrossRef]
- Hofmann, J.W.; Zhao, X.; De Cecco, M.; Peterson, A.L.; Pagliaroli, L.; Manivannan, J.; Hubbard, G.B.; Ikeno, Y.; Zhang, Y.; Feng, B.; et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell 2015, 160, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Takada, H.; Kurisaki, A. Emerging roles of nucleolar and ribosomal proteins in cancer, development, and aging. Cell Mol. Life Sci. 2015, 72, 4015–4125. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Candore, G.; Colonna-Romano, G.; Lio, D.; Caruso, M.; Hoffmann, E.; Franceschi, C.; Caruso, C. Role of Toll-like receptor 4 in acute myocardial infarction and longevity. JAMA 2004, 292, 2339–2340. [Google Scholar]
- Ramadori, G.; Ljubicic, S.; Ricci, S.; Mikropoulou, D.; Brenachot, X.; Veyrat-Durebex, C.; Aras, E.; Ioris, R.M.; Altirriba, J. S100A9 extends lifespan in insulin deficiency. Nat. Commun. 2019, 10, 3545. [Google Scholar] [CrossRef] [Green Version]
- Flynn, S.M.; Chen, C. MALT-1 mediates IL-17 neural signaling to regulate C. elegans behavior, immunity and longevity. Nat. Commun. 2020, 11, 2099. [Google Scholar] [CrossRef]
- Gruner, H.; Cortés-López, M.; Cooper, D.A.; Bauer, M.; Miura, P. CircRNA accumulation in the aging mouse brain. Sci. Rep. 2016, 6, 38907. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Lopez, M.; Gruner, M.R.; Cooper, D.A.; Gruner, H.N.; Voda, A.I.; van der Linden, A.M.; Miura, P. Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genom. 2018, 19, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, C.X.; Xue, W.; Zhang, Y.; Jiang, S.; Yin, Q.F.; Wei, J.; Yao, R.W.; Yang, L.; Chen, L.L. Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol. Cell 2017, 67, 214–227.e7. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhu, L.; Lu, C.; Wang, C.; Wang, H.; Jin, H.; Ma, X.; Cheng, Z.; Yu, C.; Wang, S.; et al. circNDUFB2 inhibits non-small cell lung cancer progression via destabilizing IGF2BPs and activating anti-tumor immunity. Nat. Commun. 2021, 12, 295. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, M.-X.; Jiang, J.-J.; Yang, L.-Q.; Yang, X.-L.; He, Y.-H.; Li, G.-H.; Kong, Q.-P. Specific Gain and Loss of Co-Expression Modules in Long-Lived Individuals Indicate a Role of circRNAs in Human Longevity. Genes 2022, 13, 749. https://doi.org/10.3390/genes13050749
Ge M-X, Jiang J-J, Yang L-Q, Yang X-L, He Y-H, Li G-H, Kong Q-P. Specific Gain and Loss of Co-Expression Modules in Long-Lived Individuals Indicate a Role of circRNAs in Human Longevity. Genes. 2022; 13(5):749. https://doi.org/10.3390/genes13050749
Chicago/Turabian StyleGe, Ming-Xia, Jian-Jun Jiang, Li-Qin Yang, Xing-Li Yang, Yong-Han He, Gong-Hua Li, and Qing-Peng Kong. 2022. "Specific Gain and Loss of Co-Expression Modules in Long-Lived Individuals Indicate a Role of circRNAs in Human Longevity" Genes 13, no. 5: 749. https://doi.org/10.3390/genes13050749
APA StyleGe, M.-X., Jiang, J.-J., Yang, L.-Q., Yang, X.-L., He, Y.-H., Li, G.-H., & Kong, Q.-P. (2022). Specific Gain and Loss of Co-Expression Modules in Long-Lived Individuals Indicate a Role of circRNAs in Human Longevity. Genes, 13(5), 749. https://doi.org/10.3390/genes13050749