
Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Subjects and Methods
2.1. Subjects
2.2. DNA Extraction and Genotyping
2.3. Statistical Analysis
3. Results
3.1. Association of Investigated SNPs with ALS Susceptibility and ALS Type
3.2. Association of Investigated SNPs with Level of Functional Impairment and Rate of Disease Progression in ALS Patients
3.3. Association of Investigated SNPs with Survival of ALS Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Ringel, S.P.; Murphy, J.R.; Alderson, M.K.; Bryan, W.; England, J.D.; Miller, R.G.; Petajan, J.H.; Smith, S.A.; Roelofs, R.I.; Ziter, F.; et al. The natural history of amyotrophic lateral sclerosis. Neurology 1993, 43, 1316. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, D.; Weishaupt, J.H. Update on amyotrophic lateral sclerosis genetics. Curr. Opin. Neurol. 2019, 32, 735–739. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- White, M.A.; Sreedharan, J. Amyotrophic lateral sclerosis: Recent genetic highlights. Curr. Opin. Neurol. 2016, 29, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.; Estrela, J. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 1847. [Google Scholar] [CrossRef]
- Harley, J.; Clarke, B.; Patani, R. The Interplay of RNA Binding Proteins, Oxidative Stress and Mitochondrial Dysfunction in ALS. Antioxidants 2021, 10, 552. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Power, J.H.T.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2008, 117, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Chang, C.-K.; Chiang, M.-H.; Toh, E.K.-W.; Chang, C.-F.; Huang, T.-H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Beal, M.F.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Fitzmaurice, P.; Shaw, I.C.; Kleiner, H.E.; Miller, R.T.; Monks, T.J.; Lau, S.S.; Mitchell, J.D.; Lynch, P.G. Evidence for DNA damage in amyotrophic lateral sclerosis. Muscle Nerve 1996, 19, 797–798. [Google Scholar] [PubMed]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.P.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Gravel, M.; Béland, L.-C.; Soucy, G.; Abdelhamid, E.; Rahimian, R.; Gravel, C.; Kriz, J. IL-10 Controls Early Microglial Phenotypes and Disease Onset in ALS Caused by Misfolded Superoxide Dismutase 1. J. Neurosci. 2016, 36, 1031–1048. [Google Scholar] [CrossRef]
- Keller, A.F.; Gravel, M.; Kriz, J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp. Neurol. 2011, 228, 69–79. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, A.; Moglia, C.; Balma, M.; Chio, A. Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: A therapeutic opportunity? CNS Neurol. Disord.-Drug Targets 2010, 9, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Winkeler, A.; Boisgard, R.; Martin, A.; Tavitian, B. Radioisotopic Imaging of Neuroinflammation: FIGURE 1. J. Nucl. Med. 2009, 51, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Zürcher, N.R.; Loggia, M.L.; Lawson, R.; Chonde, D.B.; Izquierdo-Garcia, D.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R.; Cudkowicz, M.E.; et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [11C]-PBR28. NeuroImage: Clin. 2015, 7, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Graves, M.C.; Fiala, M.; Dinglasan, L.A.V.; Liu, N.Q.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H.V. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. 2004, 5, 213–219. [Google Scholar] [CrossRef]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar]
- McCombe, P.A.; Henderson, R.D. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cao, C.; Qin, X.-Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: A meta-analysis study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Su, X.; Piao, L.; Jin, Z.; Jin, R. Involvement of Astrocytes and microRNA Dysregulation in Neurodegenerative Diseases: From Pathogenesis to Therapeutic Potential. Front. Mol. Neurosci. 2021, 14, 556215. [Google Scholar] [CrossRef]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.M.; Van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A Key Regulator of Astrocyte-Mediated Inflammatory Response. PLoS ONE 2012, 7, e44789. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2018, 56, 2137–2158. [Google Scholar] [CrossRef]
- Banack, S.A.; Dunlop, R.A.; Cox, P.A. An miRNA fingerprint using neural-enriched extracellular vesicles from blood plasma: Towards a biomarker for amyotrophic lateral sclerosis/motor neuron disease. Open Biol. 2020, 10, 200116. [Google Scholar] [CrossRef]
- Staats, K.A.; Borchelt, D.R.; Tansey, M.G.; Wymer, J. Blood-based biomarkers of inflammation in amyotrophic lateral sclerosis. Mol. Neurodegener. 2022, 17, 11. [Google Scholar] [CrossRef]
- Vrabec, K.; Koritnik, B.; Leonardis, L.; Dolenc-Grošelj, L.; Zidar, J.; Smith, B.; Vance, C.; Shaw, C.; Rogelj, B.; Glavač, D.; et al. Genetic analysis of amyotrophic lateral sclerosis in the Slovenian population. Neurobiol. Aging 2014, 36, 1601.e17–1601.e20. [Google Scholar] [CrossRef]
- Redenšek, S.; Flisar, D.; Kojović, M.; Kramberger, M.G.; Georgiev, D.; Pirtošek, Z.; Trošt, M.; Dolžan, V. Genetic variability of inflammation and oxidative stress genes does not play a major role in the occurrence of adverse events of dopaminergic treatment in Parkinson’s disease. J. Neuroinflamm. 2019, 16, 50. [Google Scholar] [CrossRef] [Green Version]
- Lyon, M.S.; Wosiski-Kuhn, M.; Gillespie, R.; Caress, J.; Milligan, C. Inflammation, Immunity, and amyotrophic lateral sclerosis: I. Etiology and pathology. Muscle Nerve 2018, 59, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomkins, J.; Banner, S.J.; McDermott, C.; Shaw, P. Mutation screening of manganese superoxide dismutase in amyotrophic lateral sclerosis. NeuroReport 2001, 12, 2319–2322. [Google Scholar] [CrossRef] [PubMed]
- Valdmanis, P.N.; Kabashi, E.; Dyck, A.; Hince, P.; Lee, J.; Dion, P.; D’Amour, M.; Souchon, F.; Bouchard, J.P.; Salachas, F.; et al. Association of paraoxonase gene cluster polymorphisms with ALS in France, Quebec, and Sweden. Neurology 2008, 71, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.B.D.S.; Santos, K.D.F.; Azevedo, R.M.; de Oliveira, R.P.D.; Leobas, A.C.D.; Bento, D.d.C.P.; Santos, R.D.S.; Reis, A.A.D.S. No association of GSTP1 rs1695 polymorphism with amyotrophic lateral sclerosis: A case-control study in the Brazilian population. PLoS ONE 2021, 16, e0247024. [Google Scholar] [CrossRef] [PubMed]
- López-López, A.; Gamez, J.; Syriani, E.; Morales, M.; Salvado, M.; Rodriguez, M.J.; Mahy, N.; Vidal-Taboada, J.M. CX3CR1 Is a Modifying Gene of Survival and Progression in Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e96528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, A.; Moglia, C.; Canosa, A.; Cammarosano, S.; Ilardi, A.; Bertuzzo, D.; Traynor, B.J.; Brunetti, M.; Barberis, M.; Mora, G.; et al. Common polymorphisms of chemokine (C-X3-C motif) receptor 1 gene modify amyotrophic lateral sclerosis outcome: A population-based study. Muscle Nerve 2017, 57, 212–216. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Walczak, M.; Lykowska-Szuber, L.; Plucinska, M.; Stawczyk-Eder, K.; Zakerska-Banaszak, O.; Eder, P.; Krela-Kazmierczak, I.; Michalak, M.; Zywicki, M.; Karlowski, W.M.; et al. Is Polymorphism in the Apoptosis and Inflammatory Pathway Genes Associated With a Primary Response to Anti-TNF Therapy in Crohn’s Disease Patients? Front. Pharmacol. 2020, 11, 1207. [Google Scholar] [CrossRef]
- Esih, K.; Goričar, K.; Rener-Primec, Z.; Dolžan, V.; Soltirovska-Šalamon, A. CARD8 and IL1B Polymorphisms Influence MRI Brain Patterns in Newborns with Hypoxic-Ischemic Encephalopathy Treated with Hypothermia. Antioxidants 2021, 10, 96. [Google Scholar] [CrossRef]
- Lee, K.A.; Gay, C.L.; Lerdal, A.; Pullinger, C.R.; Aouizerat, B.E. Cytokine polymorphisms are associated with fatigue in adults living with HIV/AIDS. Brain Behav. Immun. 2014, 40, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Ona, V.O.; Guégan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.J.; Olszewski, A.J.; Stieg, P.E.; Lee, J.-P.; Przedborski, S.; et al. Functional Role of Caspase-1 and Caspase-3 in an ALS Transgenic Mouse Model. Science 2000, 288, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Grottelli, S.; Costanzi, E.; Scarpelli, P.; Pigna, E.; Morozzi, G.; Mezzasoma, L.; Peirce, M.J.; Moresi, V.; Adamo, S.; et al. Peroxynitrite Activates the NLRP3 Inflammasome Cascade in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2017, 55, 2350–2361. [Google Scholar] [CrossRef] [Green Version]
- Olesen, M.N.; Wuolikainen, A.; Nilsson, A.C.; Wirenfeldt, M.; Forsberg, K.; Madsen, J.S.; Lillevang, S.T.; Brandslund, I.; Andersen, P.M.; Asgari, N. Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e697. [Google Scholar] [CrossRef] [Green Version]
- Polverino, A.; Rucco, R.; Stillitano, I.; Bonavita, S.; Grimaldi, M.; Minino, R.; Pesoli, M.; Trojsi, F.; D’Ursi, A.M.; Sorrentino, G.; et al. In Amyotrophic Lateral Sclerosis Blood Cytokines Are Altered, but Do Not Correlate with Changes in Brain Topology. Brain Connect. 2020, 10, 411–421. [Google Scholar] [CrossRef]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossù, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflamm. 2014, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Bresciani, G.; Cruz, I.B.M.; De Paz, J.A.; Cuevas, M.J.; González-Gallego, J. The MnSOD Ala16Val SNP: Relevance to human diseases and interaction with environmental factors. Free Radic. Res. 2013, 47, 781–792. [Google Scholar] [CrossRef]
- Sutton, A.; Imbert, A.; Igoudjil, A.; Descatoire, V.; Cazanave, S.; Pessayre, D.; Degoul, F. The manganese superoxide dismutase Ala16Val dimorphism modulates both mitochondrial import and mRNA stability. Pharm. Genom. 2005, 15, 311–319. [Google Scholar] [CrossRef]
- Ekoue, D.N.; He, C.; Diamond, A.M.; Bonini, M.G. Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim. Biophys. Acta 2017, 1858, 628–632. [Google Scholar] [CrossRef]
- Bresciani, G.; González-Gallego, J.; da Cruz, I.B.; de Paz, J.A.; Cuevas, M.J. The Ala16Val MnSOD gene polymorphism modulates oxidative response to exercise. Clin. Biochem. 2012, 46, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Tiloca, C.; Morelli, C.; Doretti, A.; Poletti, B.; Maderna, L.; Messina, S.; Gentilini, D.; Fogh, I.; Ratti, A.; et al. PON1 is a disease modifier gene in amyotrophic lateral sclerosis: Association of the Q192R polymorphism with bulbar onset and reduced survival. Neurol. Sci. 2019, 40, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, L.; Lyrenäs, L.; Morgenstern, R.; de Faire, U. A common functional C-T substitution polymorphism in the promoter region of the human catalase gene influences transcription factor binding, reporter gene transcription and is correlated to blood catalase levels. Free Radic. Biol. Med. 2001, 30, 500–505. [Google Scholar] [CrossRef]
- Esih, K.; Goričar, K.; Dolžan, V.; Rener-Primec, Z. The association between antioxidant enzyme polymorphisms and cerebral palsy after perinatal hypoxic-ischaemic encephalopathy. Eur. J. Paediatr. Neurol. 2016, 20, 704–708. [Google Scholar] [CrossRef]
- Goulas, A.; Fidani, L.; Kotsis, A.; Mirtsou, V.; Petersen, R.C.; Tangalos, E.; Hardy, J. An association study of a functional catalase gene polymorphism, −262C→T, and patients with Alzheimer’s disease. Neurosci. Lett. 2002, 330, 210–212. [Google Scholar] [CrossRef]
- Liu, K.; Liu, X.; Wang, M.; Wang, X.; Kang, H.; Lin, S.; Yang, P.; Dai, C.; Xu, P.; Li, S.; et al. Two common functional catalase gene polymorphisms (rs1001179 and rs794316) and cancer susceptibility: Evidence from 14,942 cancer cases and 43,285 controls. Oncotarget 2016, 7, 62954–62965. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-D.; Sun, Y.; Chen, N.; Huang, L.; Huang, J.-W.; Zhu, M.; Wang, T.; Ji, Y.-L. The Role of Catalase C262T Gene Polymorphism in the Susceptibility and Survival of Cancers. Sci. Rep. 2016, 6, 26973. [Google Scholar] [CrossRef] [Green Version]
- Nikolić-Kokić, A.; Stević, Z.; Blagojević, D.; Davidović-Plavšić, B.; Jones, D.R.; Spasić, M. Alterations in anti-oxidative defence enzymes in erythrocytes from sporadic amyotrophic lateral sclerosis (SALS) and familial ALS patients. Clin. Chem. Lab. Med. (CCLM) 2006, 44, 589–593. [Google Scholar] [CrossRef]
- Golenia, A.; Leśkiewicz, M.; Regulska, M.; Budziszewska, B.; Szczęsny, E.; Jagiełła, J.; Wnuk, M.; Ostrowska, M.; Lasoń, W.; Basta-Kaim, A.; et al. Catalase activity in blood fractions of patients with sporadic ALS. Pharmacol. Rep. 2014, 66, 704–707. [Google Scholar] [CrossRef]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.; Singh, R.; Kalita, J.; Misra, U. Oxidant–antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Verma, A. Clinical Manifestation and Management of Amyotrophic Lateral Sclerosis. In Amyotrophic Lateral Sclerosis; Araki, T., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Characteristic | Category/Unit | Whole Cohort | Patients with ALS-FRS-R Data |
|---|---|---|---|
| Gender | Male, N (%) | 94 (50.8) | 84 (51.2) |
| Female, N (%) | 91 (49.2) | 81 (48.8) | |
| Age at onset | Years, Median (25–75%) | 63 (56–71) [2] | 63 (57–71) |
| Age at the time of blood collection | Years, Median (25–75%) | 65 (58–72) [2] | 65 (59–73) [2] |
| ALS onset | Spinal | 133 (72.3) [1] | 121 (73.8) |
| Bulbar | 45 (24.5) | 40 (24.4) | |
| Other | 6 (3.3) | 3 (1.8) | |
| C9orf72 mutation | No, N (%) | 179 (96.8) | 159 (97.0) |
| Yes, N (%) | 6 (3.2) | 5 (3.0) | |
| Death | No, N (%) | 25 (14.0) [7] | 24 (14.9) [3] |
| Yes, N (%) | 153 (86.0) | 137 (85.1) | |
| Survival | Months, Median (25–75%) | 39.7 (26.6–68.4) [7] | 42.3 (28.0–72.3) [3] |
| Follow-up time | Months, Median (25–75%) | 104.9 (90.7–156.6) [7] | 124.2 (89.7–156.6) [3] |
| Level of functional impairment a | Points, Median (25–75%) | 34.94 (29.50–39.99) [21] | 34.94 (29.50–39.99) |
| Rate of disease progression b | Median (25–75%) | −0.80 (−1.40 to −0.30) [37] | −0.80 (−1.40 to −0.30) [16] |
| Gene | SNP | Genotype | OR (95% CI) | P | OR (95% CI)adj | Padj |
|---|---|---|---|---|---|---|
| SOD2 | rs4880 | CC | Reference | Reference | ||
| CT | 1.07 (0.69–1.66) | 0.753 | 1.01 (0.58–1.74) | 0.978 | ||
| TT | 1.06 (0.64–1.77) | 0.810 | 0.84 (0.44–1.58) | 0.586 | ||
| CT + TT | 1.07 (0.71–1.62) | 0.748 | 0.95 (0.57–1.58) | 0.834 | ||
| CAT | rs1001179 | CC | Reference | Reference | ||
| CT | 0.92 (0.63–1.35) | 0.679 | 0.94 (0.57–1.53) | 0.792 | ||
| TT | 0.84 (0.38–1.87) | 0.676 | 1.03 (0.38–2.74) | 0.958 | ||
| CT + TT | 0.91 (0.63–1.32) | 0.618 | 0.95 (0.60–1.51) | 0.826 | ||
| GPX1 | rs1050450 | CC | Reference | Reference | ||
| CT | 0.95 (0.64–1.40) | 0.788 | 1.05 (0.65–1.70) | 0.849 | ||
| TT | 0.97 (0.54–1.76) | 0.921 | 1.11 (0.51–2.40) | 0.788 | ||
| CT + TT | 0.95 (0.66–1.37) | 0.795 | 1.06 (0.67–1.67) | 0.799 | ||
| IL1B | rs1143623 | GG | Reference | Reference | ||
| GC | 0.80 (0.55–1.19) | 0.272 | 0.74 (0.46–1.21) | 0.235 | ||
| CC | 1.07 (0.57–2.01) | 0.832 | 0.93 (0.42–2.04) | 0.850 | ||
| GC + CC | 0.85 (0.59–1.23) | 0.390 | 0.78 (0.49–1.23) | 0.282 | ||
| rs16944 | TT | Reference | Reference | |||
| TC | 0.99 (0.56–1.76) | 0.979 | 0.99 (0.48–2.04) | 0.984 | ||
| CC | 1.05 (0.59–1.85) | 0.873 | 1.06 (0.52–2.17) | 0.872 | ||
| TC + CC | 1.02 (0.60–1.74) | 0.942 | 1.03 (0.52–2.01) | 0.939 | ||
| rs1071676 | GG | Reference | Reference | |||
| GC | 0.93 (0.63–1.37) | 0.714 | 1.11 (0.68–1.80) | 0.685 | ||
| CC | 0.47 (0.19–1.21) | 0.118 | 0.54 (0.18–1.61) | 0.269 | ||
| GC + CC | 0.86 (0.59–1.25) | 0.424 | 1.01 (0.63–1.60) | 0.981 | ||
| MIR146A | rs2910164 | GG | Reference | Reference | ||
| GC | 1.05 (0.72–1.55) | 0.793 | 0.85 (0.52–1.39) | 0.518 | ||
| CC | 0.74 (0.32–1.75) | 0.498 | 0.54 (0.18–1.62) | 0.273 | ||
| GC + CC | 1.01 (0.70–1.46) | 0.970 | 0.81 (0.50–1.29) | 0.364 | ||
| IL6 | rs1800795 | GG | Reference | Reference | ||
| GC | 1.18 (0.78–1.77) | 0.431 | 0.83 (0.49–1.39) | 0.467 | ||
| CC | 0.97 (0.58–1.63) | 0.914 | 1.01 (0.53–1.91) | 0.985 | ||
| GC + CC | 1.11 (0.76–1.63) | 0.584 | 0.88 (0.54–1.42) | 0.593 | ||
| TNF | rs1800629 | GG | Reference | Reference | ||
| GA | 0.88 (0.59–1.32) | 0.542 | 0.97 (0.58–1.60) | 0.897 | ||
| AA | 0.94 (0.31–2.87) | 0.916 | 0.77 (0.20–2.99) | 0.707 | ||
| GA + AA | 0.89 (0.60–1.31) | 0.548 | 0.95 (0.58–1.54) | 0.826 |
| Gene | SNP | Genotype | Spinal N (%) | Bulbar N (%) | Other N (%) | p |
|---|---|---|---|---|---|---|
| SOD2 | rs4880 | CC | 37 (80.4) | 6 (13) | 3 (6.5) | 0.153 |
| CT | 62 (68.1) | 27 (29.7) | 2 (2.2) | |||
| TT | 33 (71.7) | 12 (26.1) | 1 (2.2) | |||
| CT + TT | 95 (69.3) | 39 (28.5) | 3 (2.2) | Pdom = 0.036 | ||
| CAT | rs1001179 | CC | 81 (74.3) | 23 (21.1) | 5 (4.6) | 0.585 |
| CT | 44 (68.8) | 19 (29.7) | 1 (1.6) | |||
| TT | 7 (70) | 3 (30) | 0 (0) | |||
| CT + TT | 51 (68.9) | 22 (29.7) | 1 (1.4) | Pdom = 0.244 | ||
| GPX1 | rs1050450 | CC | 71 (76.3) | 19 (20.4) | 3 (3.2) | 0.492 |
| CT | 46 (64.8) | 22 (31) | 3 (4.2) | |||
| TT | 16 (80) | 4 (20) | 0 (0) | |||
| CT + TT | 62 (68.1) | 26 (28.6) | 3 (3.3) | Pdom = 0.446 | ||
| IL1B | rs1143623 | GG | 76 (76) | 23 (23) | 1 (1) | 0.249 |
| GC | 44 (68.8) | 16 (25) | 4 (6.3) | |||
| CC | 12 (63.2) | 6 (31.6) | 1 (5.3) | |||
| GC + CC | 56 (67.5) | 22 (26.5) | 5 (6) | Pdom = 0.128 | ||
| rs16944 | TT | 15 (62.5) | 6 (25) | 3 (12.5) | 0.063 | |
| TC | 55 (71.4) | 19 (24.7) | 3 (3.9) | |||
| CC | 62 (75.6) | 20 (24.4) | 0 (0) | |||
| TC + CC | 117 (73.6) | 39 (24.5) | 3 (1.9) | Pdom = 0.051 | ||
| rs1071676 | GG | 87 (77) | 21 (18.6) | 5 (4.4) | 0.132 | |
| GC | 41 (64.1) | 22 (34.4) | 1 (1.6) | |||
| CC | 4 (66.7) | 2 (33.3) | 0 (0) | |||
| GC + CC | 45 (64.3) | 24 (34.3) | 1 (1.4) | Pdom = 0.039 | ||
| MIR146A | rs2910164 | GG | 80 (72.1) | 28 (25.2) | 3 (2.7) | 0.665 |
| GC | 48 (73.8) | 14 (21.5) | 3 (4.6) | |||
| CC | 4 (57.1) | 3 (42.9) | 0 (0) | |||
| GC + CC | 52 (72.2) | 17 (23.6) | 3 (4.2) | Pdom = 0.872 | ||
| IL6 | rs1800795 | GG | 43 (69.4) | 17 (27.4) | 2 (3.2) | 0.655 |
| GC | 61 (70.1) | 23 (26.4) | 3 (3.4) | |||
| CC | 28 (82.4) | 5 (14.7) | 1 (2.9) | |||
| GC + CC | 89 (73.6) | 28 (23.1) | 4 (3.3) | Pdom = 0.861 | ||
| TNF | rs1800629 | GG | 91 (71.7) | 32 (25.2) | 4 (3.1) | 1.000 |
| GA | 37 (72.5) | 12 (23.5) | 2 (3.9) | |||
| AA | 4 (80) | 1 (20) | 0 (0) | |||
| GA + AA | 41 (73.2) | 13 (23.2) | 2 (3.6) | Pdom = 0.951 |
| Gene | SNP | Genotype | Level of Functional Impairment Median (25–75%) | p | Rate of Disease Progression Median (25–75%) | p |
|---|---|---|---|---|---|---|
| SOD2 | rs4880 | CC | 36.94 (31.07–40.86) | 0.199 | −0.66 (−1.25 to −0.27) | 0.455 |
| CT | 34 (29–39.6) | −0.77 (−1.47 to −0.28) | ||||
| TT | 34.18 (28.55–39.53) | −0.97 (−1.70 to −0.40) | ||||
| CT + TT | 34 (29–39.6) | Pdom = 0.073 | −0.83 (−1.50 to −0.31) | Pdom = 0.263 | ||
| CAT | rs1001179 | CC | 35 (29.21–39.98) | 0.111 | −0.81 (−1.42 to −0.34) | 0.584 |
| CT | 33.89 (29.06–39.23) | −0.71 (−1.54 to −0.28) | ||||
| TT | 39.29 (34.7–43.84) | −0.42 (−1.56 to −0.11) | ||||
| CT + TT | 34.67 (30.1–40.31) | Pdom = 0.690 | −0.67 (−1.49 to −0.27) | Pdom = 0.847 | ||
| GPX1 | rs1050450 | CC | 34.75 (30–40.71) | 0.606 | −0.66 (−1.31 to −0.33) | 0.323 |
| CT | 35 (28.62–39.17) | −0.83 (−1.50 to −0.25) | ||||
| TT | 34.51 (29.75–40.51) | −1.12 (−2.27 to −0.47) | ||||
| CT + TT | 35 (29.12–39.48) | Pdom = 0.470 | −0.92 (−1.57 to −0.26) | Pdom = 0.421 | ||
| IL1B | rs1143623 | GG | 34.02 (29.5–40) | 0.887 | −0.72 (−1.34 to −0.25) | 0.498 |
| GC | 35.14 (29–39.83) | −0.76 (−1.75 to −0.32) | ||||
| CC | 34.93 (29.66–40.82) | −0.93 (−1.54 to −0.49) | ||||
| GC + CC | 35.14 (29.38–39.83) | Pdom = 0.843 | −0.80 (−1.56 to −0.33) | Pdom = 0.307 | ||
| rs16944 | TT | 33.13 (29.57–40.3) | 0.903 | −1.04 (−1.50 to −0.53) | 0.383 | |
| TC | 34.88 (28.74–39.83) | −0.66 (−1.35 to −0.28) | ||||
| CC | 35 (30–40) | −0.82 (−1.38 to −0.21) | ||||
| TC + CC | 35 (29–40) | Pdom = 0.893 | −0.71 (−1.36 to −0.27) | Pdom = 0.174 | ||
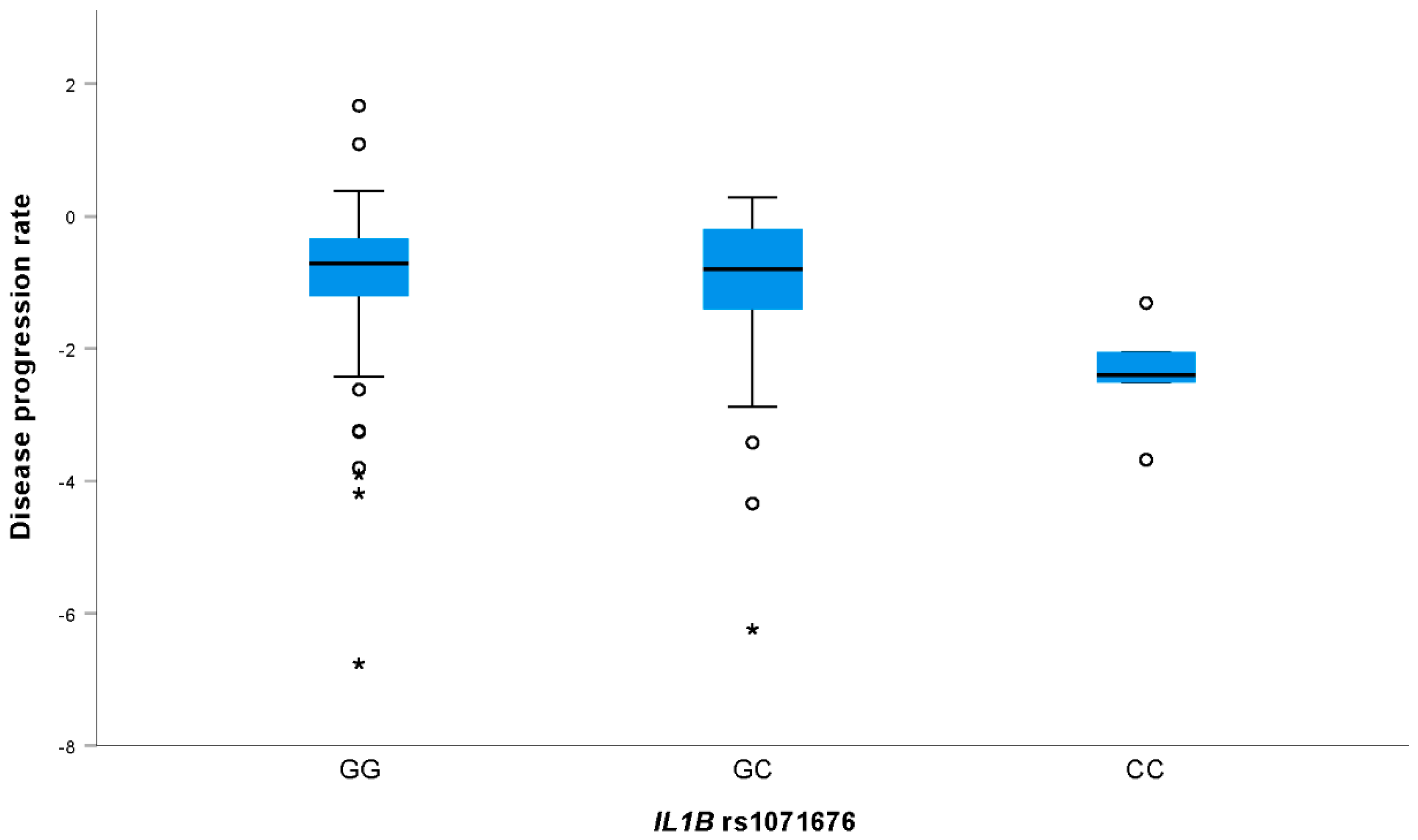
| rs1071676 | GG | 34.39 (29.86–40.06) | 0.565 | −0.71 (−1.24 to −0.34) | 0.015 | |
| GC | 34.73 (28.99–39.99) | −0.80 (−1.48 to −0.18) | ||||
| CC | 36.96 (34.52–40.12) | −2.40 (−3.10 to −1.69) | ||||
| GC + CC | 35 (29–39.98) | Pdom = 0.875 | −0.81 (−1.77 to −0.24) | Pdom = 0.690 | ||
| MIR146A | rs2910164 | GG | 34.73 (29.86–39.78) | 0.548 | −0.68 (−1.54 to −0.27) | 0.695 |
| GC | 34.38 (28.88–40.01) | −0.83 (−1.45 to −0.41) | ||||
| CC | 37.8 (31.05–41) | −0.83 (−1.17 to −0.19) | ||||
| GC + CC | 35 (29.12–40.09) | Pdom = 0.851 | −0.83 (−1.39 to −0.39) | Pdom = 0.479 | ||
| IL6 | rs1800795 | GG | 34 (30–39.6) | 0.285 | −0.53 (−1.33 to −0.18) | 0.216 |
| GC | 34 (28–39.83) | −0.83 (−1.49 to −0.37) | ||||
| CC | 36.95 (32.08–40.4) | −0.83 (−1.64 to −0.28) | ||||
| GC + CC | 34.94 (29–40.2) | Pdom = 0.634 | −0.83 (−1.52 to −0.34) | Pdom = 0.108 | ||
| TNF | rs1800629 | GG | 35.04 (30.53–40.24) | 0.512 | −0.82 (−1.39 to −0.31) | 0.827 |
| GA | 34 (27.95–37.98) | −0.67 (−2.07 to −0.27) | ||||
| AA | 38.15 (27.46–39.98) | −0.80 (−0.81 to −0.36) | ||||
| GA + AA | 34 (27.91–38.54) | Pdom = 0.272 | −0.68 (−1.49 to −0.27) | Pdom = 0.962 |
| Gene | SNP | Genotype | Median Survival (25–75%) | HR (95% CI) | p | HR (95% CI)adj | Padj |
|---|---|---|---|---|---|---|---|
| SOD2 | rs4880 | CC | 37.6 (25.7–76.0) | Reference | Reference | ||
| CT | 39.7 (27.7–69.2) | 1.00 (0.67–1.50) | 0.985 | 1.16 (0.71–1.9) | 0.558 | ||
| TT | 43.5 (23.7–63.0) | 1.20 (0.76–1.87) | 0.435 | 1.18 (0.69–2.03) | 0.549 | ||
| CT + TT | 40.7 (26.9–66.9) | 1.06 (0.73–1.55) | 0.742 | 1.17 (0.73–1.86) | 0.520 | ||
| CAT | rs1001179 | CC | 37.0 (26.6–65.8) | Reference | Reference | ||
| CT | 43.7 (26.5–72.3) | 0.83 (0.58–1.17) | 0.283 | 0.70 (0.46–1.04) | 0.079 | ||
| TT | 60.3 (56.7–85.6) | 0.61 (0.30–1.26) | 0.179 | 0.62 (0.28–1.36) | 0.231 | ||
| CT + TT | 47.6 (26.5–78.6) | 0.79 (0.57–1.1) | 0.156 | 0.68 (0.47–0.99) | 0.046 | ||
| GPX1 | rs1050450 | CC | 39.0 (28.2–69.2) | Reference | Reference | ||
| CT | 39.7 (25.3–72.8) | 0.89 (0.63–1.26) | 0.507 | 0.85 (0.58–1.24) | 0.396 | ||
| TT | 40.5 (29.0–65.8) | 0.99 (0.58–1.68) | 0.969 | 1.06 (0.53–2.14) | 0.869 | ||
| CT + TT | 39.7 (25.3–67.6) | 0.91 (0.66–1.26) | 0.570 | 0.88 (0.61–1.26) | 0.477 | ||
| IL1B | rs1143623 | GG | 41.8 (26.5–74.5) | Reference | Reference | ||
| GC | 39.0 (25.3–68.4) | 1.12 (0.8–1.58) | 0.503 | 1.21 (0.82–1.78) | 0.332 | ||
| CC | 37.0 (34.0–56.7) | 1.32 (0.76–2.31) | 0.322 | 0.78 (0.38–1.59) | 0.491 | ||
| GC + CC | 39.0 (27.7–67.6) | 1.16 (0.84–1.6) | 0.361 | 1.11 (0.77–1.6) | 0.572 | ||
| rs16944 | TT | 37.0 (33.2–56.7) | Reference | Reference | |||
| TC | 43.7 (25.7–76.0) | 0.66 (0.4–1.11) | 0.118 | 1.08 (0.55–2.09) | 0.827 | ||
| CC | 39.4 (26.5–66.9) | 0.74 (0.45–1.23) | 0.245 | 1.12 (0.59–2.13) | 0.722 | ||
| TC + CC | 41.8 (26.5–72.3) | 0.70 (0.44–1.14) | 0.150 | 1.1 (0.59–2.06) | 0.754 | ||
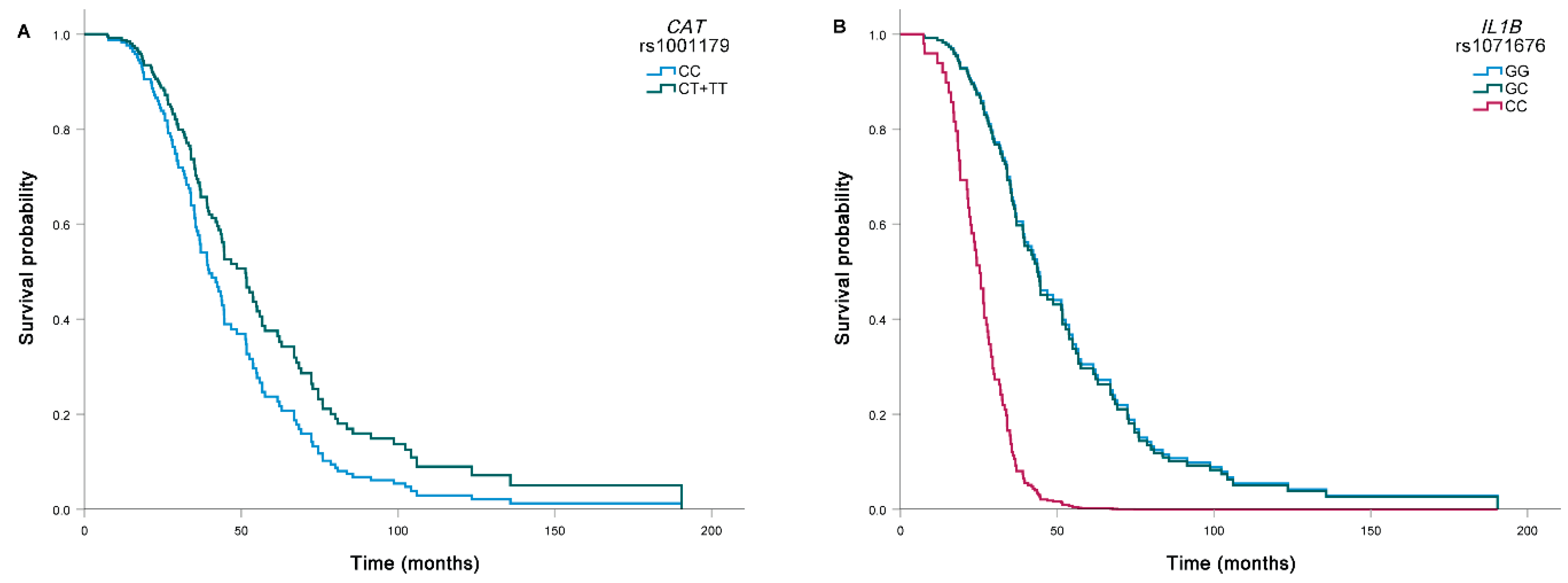
| rs1071676 | GG | 39.7 (25.7–66.9) | Reference | Reference | |||
| GC | 41.8 (28.7–83.6) | 0.75 (0.53–1.07) | 0.110 | 1.03 (0.69–1.53) | 0.900 | ||
| CC | 24.3 (18.4–32.0) | 1.53 (0.62–3.77) | 0.358 | 5.02 (1.92–13.16) | 0.001 | ||
| GC + CC | 40.5 (28.0–78.6) | 0.79 (0.57–1.11) | 0.171 | 1.13 (0.77–1.66) | 0.529 | ||
| MIR146A | rs2910164 | GG | 42.9 (25.7–72.3) | Reference | Reference | ||
| GC | 39.0 (27.7–65.8) | 1.03 (0.73–1.45) | 0.860 | 1 (0.68–1.46) | 0.989 | ||
| CC | 36.7 (31.4–60.3) | 1.26 (0.55–2.88) | 0.584 | 1.42 (0.57–3.54) | 0.448 | ||
| GC + CC | 39.0 (27.7–65.8) | 1.05 (0.76–1.46) | 0.768 | 1.03 (0.71–1.49) | 0.876 | ||
| IL6 | rs1800795 | GG | 37.0 (26.6–78.6) | Reference | Reference | ||
| GC | 39.7 (25.7–60.3) | 1.02 (0.84–1.73) | 0.317 | 1.06 (0.69–1.62) | 0.793 | ||
| CC | 40.7 (29.9–74.5) | 0.90 (0.56–1.43) | 0.647 | 0.84 (0.48–1.46) | 0.527 | ||
| GC + CC | 40.5 (26.5–66.9) | 1.10 (0.78–1.55) | 0.585 | 1 (0.66–1.5) | 0.984 | ||
| TNF | rs1800629 | GG | 39.0 (25.7–67.6) | Reference | Reference | ||
| GA | 41.8 (29.8–74.5) | 0.92 (0.64–1.33) | 0.670 | 0.78 (0.5–1.21) | 0.266 | ||
| AA | 37.0 (32.5–57.6) | 1.25 (0.51–3.06) | 0.631 | 1.45 (0.58–3.58) | 0.426 | ||
| GA + AA | 40.7 (29.8–74.5) | 0.95 (0.67–1.35) | 0.778 | 0.84 (0.56–1.27) | 0.411 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravnik-Glavač, M.; Goričar, K.; Vogrinc, D.; Koritnik, B.; Lavrenčič, J.G.; Glavač, D.; Dolžan, V. Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis. Genes 2022, 13, 757. https://doi.org/10.3390/genes13050757
Ravnik-Glavač M, Goričar K, Vogrinc D, Koritnik B, Lavrenčič JG, Glavač D, Dolžan V. Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis. Genes. 2022; 13(5):757. https://doi.org/10.3390/genes13050757
Chicago/Turabian StyleRavnik-Glavač, Metka, Katja Goričar, David Vogrinc, Blaž Koritnik, Jakob Gašper Lavrenčič, Damjan Glavač, and Vita Dolžan. 2022. "Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis" Genes 13, no. 5: 757. https://doi.org/10.3390/genes13050757
APA StyleRavnik-Glavač, M., Goričar, K., Vogrinc, D., Koritnik, B., Lavrenčič, J. G., Glavač, D., & Dolžan, V. (2022). Genetic Variability of Inflammation and Oxidative Stress Genes Affects Onset, Progression of the Disease and Survival of Patients with Amyotrophic Lateral Sclerosis. Genes, 13(5), 757. https://doi.org/10.3390/genes13050757