
A Common Missense Variant Causing Factor XI Deficiency and Increased Bleeding Tendency in Maine Coon Cats

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Samples
2.2. Coagulation Assays
2.3. DNA Sequencing
2.4. Genotyping
2.5. Factor XI Immunoprecipitation from Plasma
2.6. Recombinant FXI
2.7. Factor XI Activity in aPTT Assays
2.8. Factor XI Activation
2.9. Factor IX Activation by FXIa
2.10. Chromogenic Assay for FXIa Activity
2.11. Statistical Analysis
3. Results
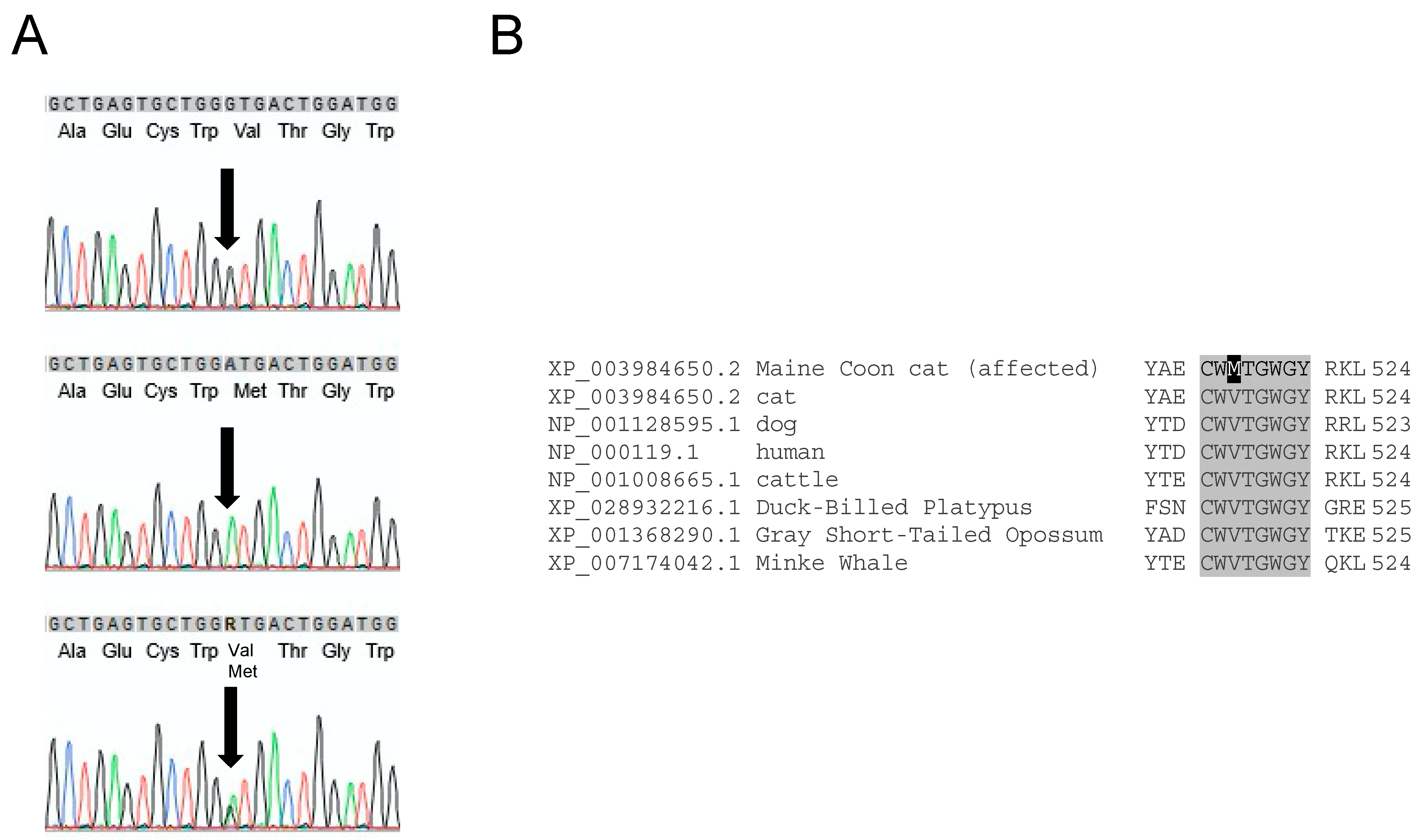
3.1. F11 Sequence Variants in Maine Coon Cats
3.2. Genotyping for FXI-V516M
3.3. Clinical Signs
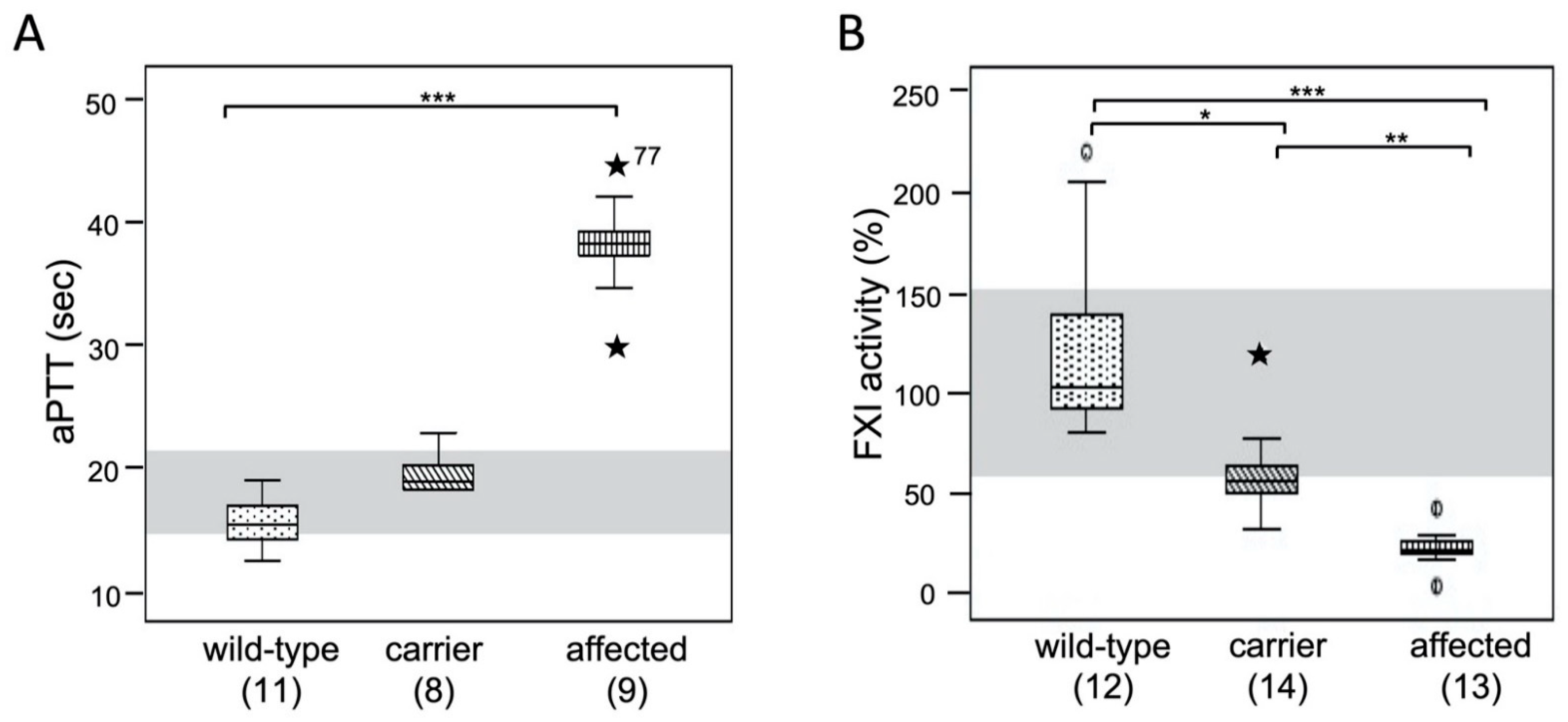
3.4. Coagulation Test Results: aPTT and FXI Activity
3.5. Factor XI in Feline Plasma
3.6. Factor XI Activity
3.7. Factor XI Activation
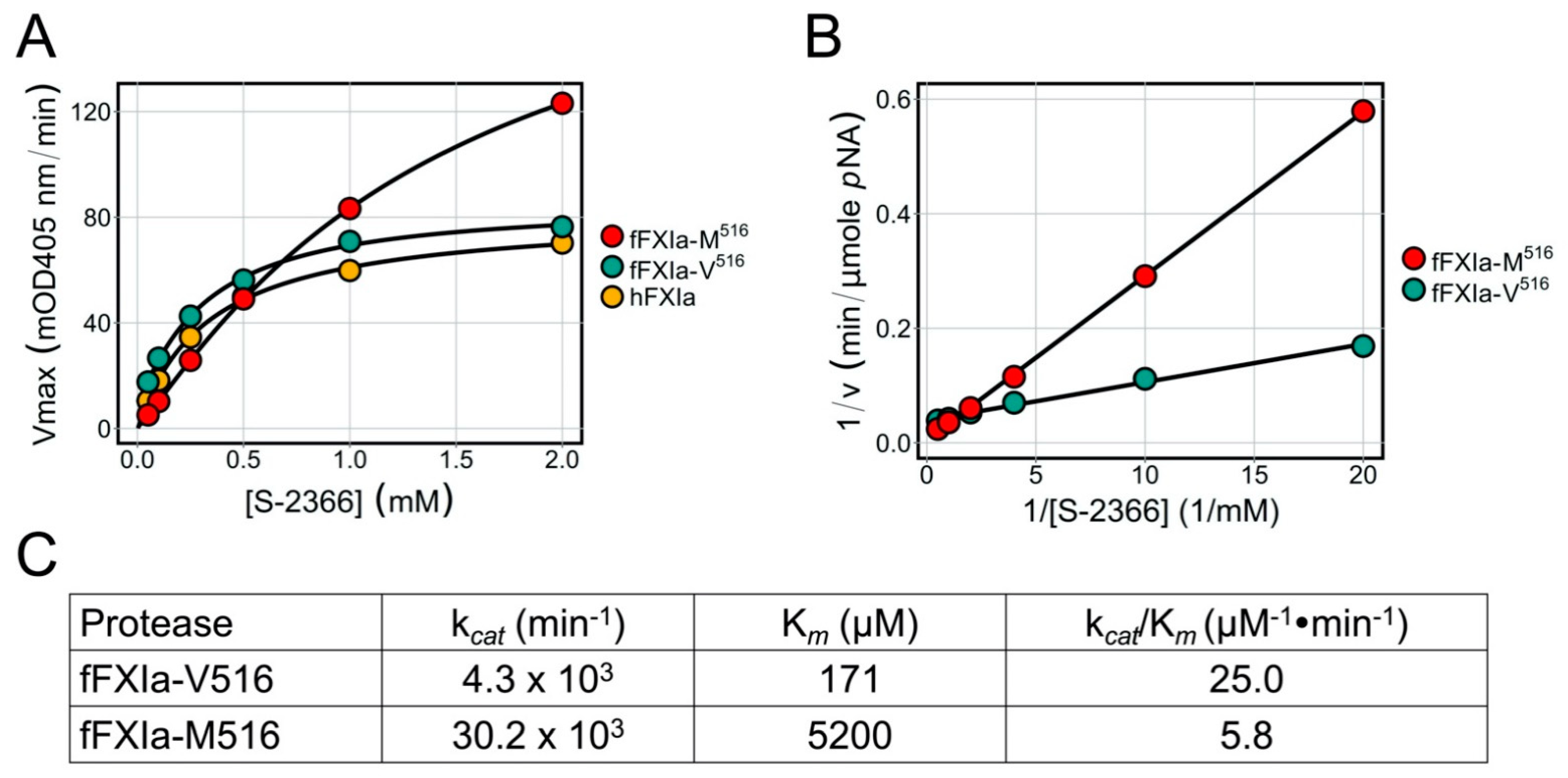
3.8. Factor XIa Activity in a Chromogenic Assay
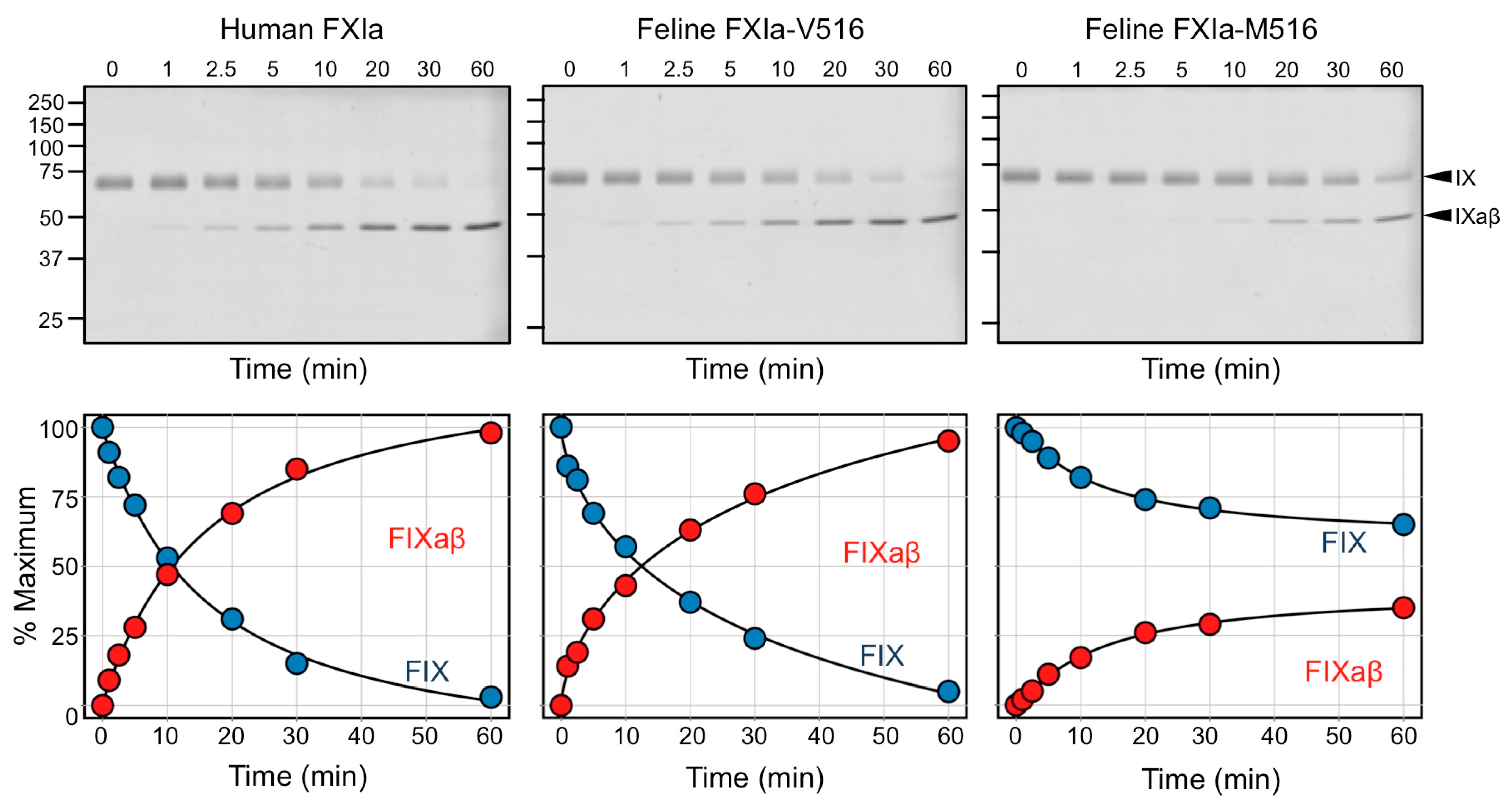
3.9. Factor IX Activation by FXIa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ponczek, M.B.; Shamanaev, A.; LaPlace, A.; Dickeson, S.K.; Srivastava, P.; Sun, M.; Gruber, A.; Kastrup, C.; Emsley, J.; Gailani, D. The Evolution of Factor XI and the Kallikrein-Kinin System. Blood Adv. 2020, 4, 6135–6147. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, B.M.; Matafonov, A.; Ivanov, I.; Sun, M.; Cheng, Q.; Dickeson, S.K.; Li, C.; Sun, D.; Verhamme, I.M.; Emsley, J.; et al. An Update on Factor XI Structure and Function. Thromb. Res. 2018, 161, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.E.; Mandle, R.; Kaplan, A.P. Association of Factor XI and High Molecular Weight Kininogen in Human Plasma. J. Clin. Investig. 1977, 60, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.L.; Dreskin, O.H.; Rosenthal, N. Plasma Thromboplastin Antecedent (PTA) Deficiency: Clinical, Coagulation, Therapeutic and Hereditary Aspects of a New Hemophilia-like Disease. Blood 1955, 10, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Duga, S.; Salomon, O. Congenital Factor XI Deficiency: An Update. Semin. Thromb. Hemost. 2013, 39, 621–631. [Google Scholar] [CrossRef]
- Santoro, C.; Mauro, R.D.; Baldacci, E.; Angelis, F.D.; Abbruzzese, R.; Barone, F.; Bochicchio, R.A.; Ferrara, G.; Guarini, A.; Foà, R.; et al. Bleeding Phenotype and Correlation with Factor XI (FXI) Activity in Congenital FXI Deficiency: Results of a Retrospective Study from a Single Centre. Haemophilia 2015, 21, 496–501. [Google Scholar] [CrossRef]
- James, P.; Salomon, O.; Mikovic, D.; Peyvandi, F. Rare Bleeding Disorders—Bleeding Assessment Tools, Laboratory Aspects and Phenotype and Therapy of FXI Deficiency. Haemophilia 2014, 20, 71–75. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B. Factor XI Deficiency—Resolving the Enigma? Hematology 2009, 2009, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Mumford, A.D.; Ackroyd, S.; Alikhan, R.; Bowles, L.; Chowdary, P.; Grainger, J.; Mainwaring, J.; Mathias, M.; O’Connell, N. Guideline for the Diagnosis and Management of the Rare Coagulation Disorders. Br. J. Haematol. 2014, 167, 304–326. [Google Scholar] [CrossRef]
- Asakai, R.; Chung, D.W.; Davie, E.W.; Seligsohn, U. Factor XI Deficiency in Ashkenazi Jews in Israel. N. Engl. J. Med. 1991, 325, 153–158. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Rimoldi, V.; Menegatti, M.; Peyvandi, F.; Salomon, O.; Duga, S. Exploring the Global Landscape of Genetic Variation in Coagulation Factor XI Deficiency. Blood 2017, 130, e1–e6. [Google Scholar] [CrossRef] [Green Version]
- Shpilberg, O.; Peretz, H.; Zivelin, A.; Yatuv, R.; Chetrit, A.; Kulka, T.; Stern, C.; Weiss, E.; Seligsohn, U. One of the Two Common Mutations Causing Factor XI Deficiency in Ashkenazi Jews (Type II) Is Also Prevalent in Iraqi Jews, Who Represent the Ancient Gene Pool of Jews. Blood 1995, 85, 429–432. [Google Scholar] [CrossRef] [Green Version]
- Peretz, H.; Mulai, A.; Usher, S.; Zivelin, A.; Segal, A.; Weisman, Z.; Mittelman, M.; Lupo, H.; Lanir, N.; Brenner, B.; et al. The Two Common Mutations Causing Factor XI Deficiency in Jews Stem from Distinct Founders: One of Ancient Middle Eastern Origin and Another of More Recent European Origin. Blood 1997, 90, 2654–2659. [Google Scholar] [CrossRef]
- Zivelin, A.; Bauduer, F.; Ducout, L.; Peretz, H.; Rosenberg, N.; Yatuv, R.; Seligsohn, U. Factor XI Deficiency in French Basques Is Caused Predominantly by an Ancestral Cys38Arg Mutation in the Factor XI Gene. Blood 2002, 99, 2448–2454. [Google Scholar] [CrossRef] [Green Version]
- Tiscia, G.L.; Favuzzi, G.; Lupone, M.R.; Cappucci, F.; Schiavulli, M.; Mirabelli, V.; D’Andrea, G.; Chinni, E.; Giuliani, N.; Caliandro, R.; et al. Factor XI Gene Variants in Factor XI-Deficient Patients of Southern Italy: Identification of a Novel Mutation and Genotype–Phenotype Relationship. Hum. Genome Var. 2017, 4, 17043. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Peretz, H.; Butler, R.; Mountford, R.; Keeney, S.; Zacharski, L.; Zivelin, A.; Seligsohn, U. A Common Ancestral Mutation (C128X) Occurring in 11 Non-Jewish Families from the UK with Factor XI Deficiency. J. Thromb. Haemost. 2004, 2, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.; Laor, A.; Lupo, H.; Zivelin, A.; Lanir, N.; Seligsohn, U. Bleeding Predictors in Factor-XI-Deficient Patients. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 1997, 8, 511–515. [Google Scholar] [CrossRef]
- Marron, B.M.; Robinson, J.L.; Gentry, P.A.; Beever, J.E. Identification of a Mutation Associated with Factor XI Deficiency in Holstein Cattle. Anim. Genet. 2004, 35, 454–456. [Google Scholar] [CrossRef]
- Gentry, P.A.; Ross, M.L. Coagulation Factor XI Deficiency in Holstein Cattle: Expression and Distribution of Factor XI Activity. Can. J. Vet. Res. 1994, 58, 242–247. [Google Scholar]
- Meydan, H.; Yildiz, M.A.; Özdil, F.; Gedik, Y.; Özbeyaz, C. Identification of Factor XI Deficiency in Holstein Cattle in Turkey. Acta Vet. Scand. 2009, 51, 5. [Google Scholar] [CrossRef] [Green Version]
- Kunieda, M.; Tsuji, T.; Abbasi, A.R.; Khalaj, M.; Ikeda, M.; Miyadera, K.; Ogawa, H.; Kunieda, T. An Insertion Mutation of the Bovine F11 Gene Is Responsible for Factor XI Deficiency in Japanese Black Cattle. Mamm. Genome 2005, 16, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Mondal, K.; Chakravarti, S.; Ghosh, A.K.; Kumar, S.; Nayak, B.; Nandi, S.; Sarkar, U.; Deb, R.; De, A.; Biswas, J. Novel Identification of Factor XI Deficiency in Indian Sahiwal (Bos Indicus) Cattle. Mol. Biol. Rep. 2016, 43, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Knowler, C.; Giger, U.; Dodds, W.J.; Brooks, M. Factor XI Deficiency in Kerry Blue Terriers. J. Am. Vet. Med. Assoc. 1994, 205, 1557–1561. [Google Scholar] [PubMed]
- Tcherneva, E.; Giger, U. Molecular Base of Coagulation Factor XI Deficiency in Kerry Blue Terrier. Bulg. J. Vet. Med. 2007, 10, 247–255. [Google Scholar]
- Maruyama, H.; Brooks, M.B.; Stablein, A.; Frye, A. Factor XII Deficiency Is Common in Domestic Cats and Associated with Two High Frequency F12 Mutations. Gene 2019, 706, 6–12. [Google Scholar] [CrossRef]
- Stokol, T.; Brooks, M.B.; Erb, H.N. Effect of Citrate Concentration on Coagulation Test Results in Dogs. J. Am. Vet. Med. Assoc. 2000, 217, 1672–1677. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, R.J.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Tucker, E.I.; Pine, M.S.; Sisler, I.; Matafonov, A.; Sun, M.; White-Adams, T.C.; Smith, S.A.; Hanson, S.R.; McCarty, O.J.T.; et al. A Role for Factor XIIa–Mediated Factor XI Activation in Thrombus Formation in Vivo. Blood 2010, 116, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Verhamme, I.M.; Messer, A.; Sun, M.; Smith, S.B.; Bajaj, S.P.; Gailani, D. A Sequential Mechanism for Exosite-Mediated Factor IX Activation by Factor XIa. J. Biol. Chem. 2012, 287, 38200–38209. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Gailani, D. Identification of a Factor IX Binding Site on the Third Apple Domain of Activated Factor XI. J. Biol. Chem. 1996, 271, 29023–29028. [Google Scholar] [CrossRef] [Green Version]
- Kruskal, W.H.; Wallis, W.A. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Bajaj, S.P.; Birktoft, J.J. [6] Human Factor IX and Factor IXa. In Methods in Enzymology; Proteolytic Enzymes in Coagulation, Fibrinolysis, and Complement Activation Part A: Mammalian Blood Coagulation Factors and Inhibitors; Academic Press: Cambridge, MA, USA, 1993; Volume 222, pp. 96–128. [Google Scholar]
- McVey, J.H.; Rallapalli, P.M.; Kemball-Cook, G.; Hampshire, D.J.; Giansily-Blaizot, M.; Gomez, K.; Perkins, S.J.; Ludlam, C.A. The European Association for Haemophilia and Allied Disorders (EAHAD) Coagulation Factor Variant Databases: Important Resources for Haemostasis Clinicians and Researchers. Haemophilia 2020, 26, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Smith, S.A.; Morrissey, J.H. Polyphosphate Is a Cofactor for the Activation of Factor XI by Thrombin. Blood 2011, 118, 6963–6970. [Google Scholar] [CrossRef] [Green Version]
- Kearney, K.J.; Butler, J.; Posada, O.M.; Wilson, C.; Heal, S.; Ali, M.; Hardy, L.; Ahnström, J.; Gailani, D.; Foster, R.; et al. Kallikrein Directly Interacts with and Activates Factor IX, Resulting in Thrombin Generation and Fibrin Formation Independent of Factor XI. Proc. Natl. Acad. Sci. USA 2021, 118, e2014810118. [Google Scholar] [CrossRef]
- Nichols, T.C.; Hough, C.; Agerso, H.; Ezban, M.; Lillicrap, D. Canine Models of Inherited Bleeding Disorders in the Development of Coagulation Assays, Novel Protein Replacement and Gene Therapies. J. Thromb. Haemost. 2016, 14, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Giansily-Blaizot, M.; Rallapalli, P.M.; Perkins, S.J.; Kemball-Cook, G.; Hampshire, D.J.; Gomez, K.; Ludlam, C.A.; McVey, J.H. The EAHAD Blood Coagulation Factor VII Variant Database. Hum. Mutat. 2020, 41, 1209–1219. [Google Scholar] [CrossRef]
- Kwon, M.-J.; Kim, H.-J.; Bang, S.-H.; Kim, S.-H. Severe Factor XI Deficiency in a Korean Woman with a Novel Missense Mutation (Val498Met) and Duplication G Mutation in Exon 13 of the F11 Gene. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2008, 19, 679–683. [Google Scholar] [CrossRef]
- McMullen, B.A.; Fujikawa, K.; Davie, E.W. Location of the Disulfide Bonds in Human Coagulation Factor XI: The Presence of Tandem Apple Domains. Biochemistry 1991, 30, 2056–2060. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Chung, D.W.; Hendrickson, L.E.; Davie, E.W. Amino Acid Sequence of Human Factor XI, a Blood Coagulation Factor with Four Tandem Repeats That Are Highly Homologous with Plasma Prekallikrein. Biochemistry 1986, 25, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Emsley, J.; McEwan, P.A.; Gailani, D. Structure and Function of Factor XI. Blood 2010, 115, 2569–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouma, B.N.; Meijers, J.C.M. Role of Blood Coagulation Factor XI in Downregulation of Fibrinolysis. Curr. Opin. Hematol. 2000, 7, 266–272. [Google Scholar] [CrossRef]
- Mohammed, B.M.; Monroe, D.M.; Gailani, D. Mouse Models of Hemostasis. Platelets 2020, 31, 417–422. [Google Scholar] [CrossRef]
- Gailani, D.; Bane, C.E.; Gruber, A. Factor XI and Contact Activation as Targets for Antithrombotic Therapy. J. Thromb. Haemost. 2015, 13, 1383–1395. [Google Scholar] [CrossRef] [Green Version]
- Gentry, P.A.; Black, W.D. Prevalence and Inheritance of Factor XI (Plasma Thromboplastin Antecedent) Deficiency in Cattle. J. Dairy Sci. 1980, 63, 616–620. [Google Scholar] [CrossRef]
- Troxel, M.T.; Brooks, M.B.; Esterline, M.L. Congenital Factor XI Deficiency in a Domestic Shorthair Cat. J. Am. Anim. Hosp. Assoc. 2002, 38, 549–553. [Google Scholar] [CrossRef]
- O’Halloran, C.; Cerna, P.; Breheny, C.; Reed, N.; Rolph, K.; Cade, S.; Jones, J.; Brown, R.A.L.; Slade, S.; Papasouliotis, K.; et al. Investigation of Pathological Haemorrhage in Maine Coon Cats. Vet. Rec. 2020, 187, e75. [Google Scholar] [CrossRef]
- Salomon, O.; Gailani, D. A Proposal for Managing Bleeding in Patients on Therapeutic Factor XI(a) Inhibitors. J. Thromb. Haemost. 2022, 20, 32–38. [Google Scholar] [CrossRef]
- Birkbeck, R.; Humm, K.; Cortellini, S. A Review of Hyperfibrinolysis in Cats and Dogs. J. Small Anim. Pract. 2019, 60, 641–655. [Google Scholar] [CrossRef]
- Osekavage, K.E.; Brainard, B.M.; Lane, S.L.; Almoslem, M.; Arnold, R.D.; Koenig, A. Pharmacokinetics of Tranexamic Acid in Healthy Dogs and Assessment of Its Antifibrinolytic Properties in Canine Blood. Am. J. Vet. Res. 2018, 79, 1057–1063. [Google Scholar] [CrossRef]
- Marín, L.M.; Iazbik, M.C.; Zaldivar-Lopez, S.; Guillaumin, J.; McLoughlin, M.A.; Couto, C.G. Epsilon Aminocaproic Acid for the Prevention of Delayed Postoperative Bleeding in Retired Racing Greyhounds Undergoing Gonadectomy. Vet. Surg. VS 2012, 41, 594–603. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cats | Total Number # | F11 Genotype (XP_003984650.1:p.V516M) | Mutant (M516) Allele Frequency | ||
|---|---|---|---|---|---|
| VV | VM | MM | |||
| MCCs from USA | 39 | 12 | 14 | 13 | 0.51 |
| MCCs Europe | 263 | 152 | 100 | 11 | 0.23 |
| Other Feline Breeds * | 100 | 100 | - | - | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuder, H.; Dickeson, S.K.; Brooks, M.B.; Kehl, A.; Müller, E.; Gailani, D.; Giger, U. A Common Missense Variant Causing Factor XI Deficiency and Increased Bleeding Tendency in Maine Coon Cats. Genes 2022, 13, 792. https://doi.org/10.3390/genes13050792
Kuder H, Dickeson SK, Brooks MB, Kehl A, Müller E, Gailani D, Giger U. A Common Missense Variant Causing Factor XI Deficiency and Increased Bleeding Tendency in Maine Coon Cats. Genes. 2022; 13(5):792. https://doi.org/10.3390/genes13050792
Chicago/Turabian StyleKuder, Henrike, S. Kent Dickeson, Marjory B. Brooks, Alexandra Kehl, Elisabeth Müller, David Gailani, and Urs Giger. 2022. "A Common Missense Variant Causing Factor XI Deficiency and Increased Bleeding Tendency in Maine Coon Cats" Genes 13, no. 5: 792. https://doi.org/10.3390/genes13050792