
Genetic and Epigenetic Signatures Associated with the Divergence of Aquilegia Species

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction and Whole-Genome Sequencing
2.2. Sequence Assembly, Functional Annotation and Genetic Diversity
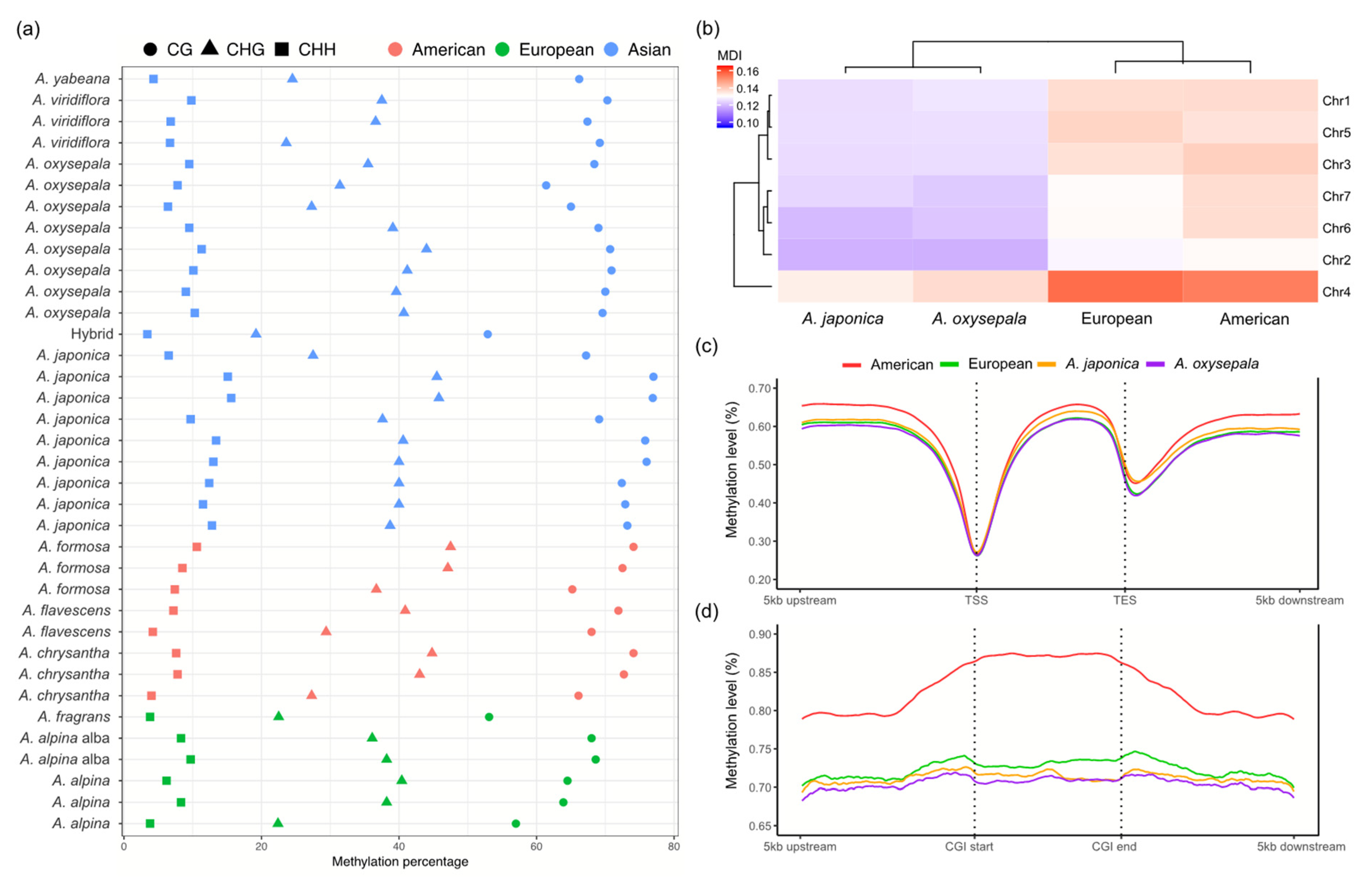
2.3. Cytosine Methylation Pattern and Epigenetic Population Structure
2.4. Associations between the Genetic Variation and Cytosine Methylation
2.5. Identification of Conservative Clade-Specific Variant
2.6. Functional Analysis
3. Results
3.1. Population Structure and Nucleotide Variation Pattern
3.2. Identification of the Genomic Regions Indicating Selection Pressure and Highly Impactful Genetic Variations
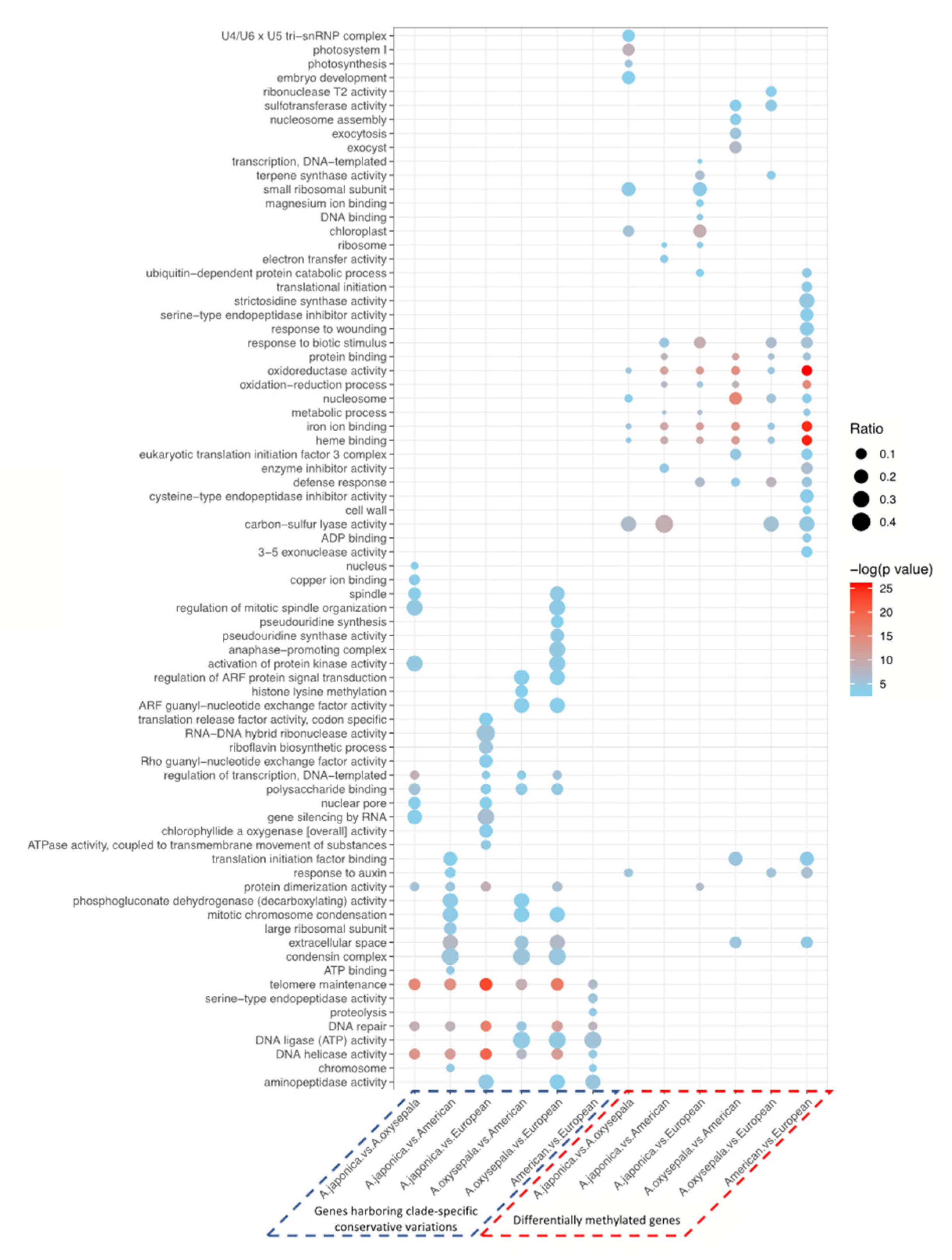
3.3. Cytosine Methylation Patterns and Differentially Methylated Genes
3.4. Association between Epigenetic Genetic and Variation
4. Discussion
4.1. Genetically Determined Mechanisms Associated with the Rapid Diversification of Aquilegia Species
4.2. Associations between Cytosine Methylation and Diversification of Aquilegia Species
4.3. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schluter, D. Ecological causes of adaptive radiation. Am. Nat. 1996, 148, S40–S64. [Google Scholar] [CrossRef]
- Schluter, D. The Ecology of Adaptive Radiation. Heredity 2000, 86, 749–750. [Google Scholar]
- Givnish, T.J. Adaptive Radiation and Molecular Systematics: Aims and Conceptual Issues; Molecular Evolution and Adaptive Radiation; Cambridge University Press: Cambridge, UK, 1997; pp. 1–54. [Google Scholar]
- Givnish, T.J. Adaptive radiation versus ‘radiation’ and ‘explosive diversification’: Why conceptual distinctions are fundamental to understanding evolution. New Phytol. 2015, 207, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.G.; Olson, E.C. The major features of evolution. Q. Rev. Biol. 1953, 8, 87–88. [Google Scholar]
- Losos, J.B. Adaptive radiation, ecological opportunity, and evolutionary determinism. American Society of Naturalists E. O. Wilson award address. Am. Nat. 2010, 175, 623–639. [Google Scholar] [CrossRef]
- Carlquist Motley, T.J. Tarweeds and Silverswords, Evolution of the Madiinae (Asteraceae). Econ. Bot. 2003, 58, 123–124. [Google Scholar] [CrossRef]
- Losos, J.B.; Ricklefs, R.E. Adaptation and diversification on islands. Nature 2009, 457, 830–836. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Berglund, J.; Almén, M.S.; Maqbool, K.; Grabherr, M.; Martinez-Barrio, A.; Promerová, M.; Rubin, C.; Wang, C.; Zamani, N.; et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 2015, 518, 371–375. [Google Scholar] [CrossRef]
- Irisarri, I.; Singh, P.; Koblmüller, S.; Torres-Dowdall, J.; Henning, F.; Franchini, P.; Fischer, C.; Lemmon, A.R.; Lemmon, E.M.; Thallinger, G.G.; et al. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat. Commun. 2018, 9, 3159–3171. [Google Scholar] [CrossRef]
- Wagner, C.E.; Harmon, L.J.; Seehausen, O. Ecological opportunity and sexual selection together predict adaptive radiation. Nature 2012, 487, 366–369. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Berner, D.; Salzburger, W. The genomics of organismal diversification illuminated by adaptive radiations. Trends Genet. 2015, 31, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Brawand, D.; Wagner, C.; Li, Y.; Malinsky, M.; Keller, I.; Fan, S.; Simakov, O.; Ng, A.; Lim, Z.; Bezault, E.; et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature 2014, 513, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, A.G.P.; Rüber, L.; Newton, J.; DasMahapatra, K.K.; Balarin, J.D.; Bruun, K.; Day, J. Niche divergence facilitated by fine-scale ecological partitioning in a recent cichlid fish adaptive radiation. Evolution 2016, 70, 2718–2735. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Barton, N.H.; Charlesworth, B. The Sources of Adaptive Variation. Proc. R. Soc. B Biol. Sci. 2017, 284, 20162864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, G.; Heard, E. Advances in Epigenetics Link Genetics to the Environment and Disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Boquete, M.T.; Muyle, A.; Alonso, C. Plant Epigenetics: Phenotypic and Functional Diversity beyond the DNA Sequence. Am. J. Bot. 2021, 108, 553–558. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Fior, S.; Li, M.; Oxelman, B.; Viola, R.; Hodges, S.A.; Ometto, L.; Varotto, C. Spatiotemporal reconstruction of the Aquilegia rapid radiation through next-generation sequencing of rapidly evolving cpDNA regions. New Phytol. 2013, 198, 579–592. [Google Scholar] [CrossRef]
- Filiault, D.L.; Ballerini, E.S.; Mandáková, T.; Aköz, G.; Derieg, N.J.; Schmutz, J.; Jenkins, J.; Grimwood, J.; Shu, S.; Hayes, R.D.; et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLife 2018, 7, e36436. [Google Scholar] [CrossRef] [PubMed]
- Munz, P.A. Aquilegia: The cultivated and wild columbines. Gentes Herbarum 1946, 7, 1–150. [Google Scholar]
- Bastida, J.M.; Herrera, C.M. Extended phylogeny of Aquilegia: The biogeographical and ecological patterns of two simultaneous but contrasting radiations. Plant Syst. Evol. 2010, 284, 171–185. [Google Scholar] [CrossRef]
- Whittall, J.B.; Hodges, S.A. Pollinator shifts drive increasingly long nectar spurs in columbine flowers. Nature 2007, 447, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Alcántara, J.M.; Bastida, J.M.; Rey, P.J. Linking divergent selection on vegetative traits to environmental variation and phenotypic diversification in the Iberian columbines (Aquilegia). J. Evol. Biol. 2010, 23, 1218–1233. [Google Scholar] [CrossRef]
- Kramer, E.M.; Hodges, S.A. Aquilegia as a model system for the evolution and ecology of petals. Philos. Trans. Biol. Sci. 2010, 365, 477–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, B.; Yant, L.; Hodges, S.A.; Kramer, E.M. Understanding the development and evolution of novel floral form in Aquilegia. Curr. Opin. Plant Biol. 2014, 17, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.L.; Alcantara, J.; Rey, P.J.; Medrano, M.; Guitián, J.; Castellanos, M.C.; Bastida, J.M.; Jaime, R.; Herrera, C.M. Geographic genetic structure of Iberian columbines (gen. Aquilegia). Plant Syst. Evol. 2017, 303, 1145–1160. [Google Scholar] [CrossRef]
- Tang, L.L.; Yu, Q.; Sun, J.F.; Huang, S.Q. Floral traits and isolation of three sympatric Aquilegia species in the Qinling Mountains, China. Plant Syst. Evol. 2007, 267, 121–128. [Google Scholar] [CrossRef]
- Li, L.F.; Wang, H.Y.; Pang, D.; Liu, Y.; Liu, B.; Xiao, H.X. Phenotypic and genetic evidence for ecological speciation of Aquilegia japonica and A. oxysepala. New Phytol. 2014, 204, 1028–1040. [Google Scholar] [CrossRef]
- Li, L.F.; Pang, D.; Liao, Q.L.; Xiao, H.X. Genomic and EST microsatellite markers for Aquilegia flabellata and cross-amplification in A. oxysepala (Ranunculaceae). Am. J. Bot. 2011, 98, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.E.; Johnson, K.C.; Skinner, L.; Koestler, D.C.; Christensen, B.C. Epigenetic and genetic burden measures are associated with tumor characteristics in invasive breast carcinoma. Epigenetics 2016, 11, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; David, R. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Dawe, R.K. Mechanisms of plant spindle formation. Chromosome Res. 2011, 19, 335–344. [Google Scholar] [CrossRef]
- Aguirre-Portoles, C.; Bird, A.W.; Hyman, A.; Canamero, M.; de Castro, I.P.; Malunnbres, M. Tpx2 Controls Spindle Integrity, Genome Stability, and Tumor Development. Cancer Res. 2012, 72, 1518–1528. [Google Scholar] [CrossRef] [Green Version]
- Fukui, K.; Nakagawa, N.; Kitamura, Y.; Nishida, Y.; Masui, R.; Kuramitsu, S. Crystal Structure of MutS2 Endonuclease Domain and the Mechanism of Homologous Recombination Suppression. J. Biol. Chem. 2008, 283, 33417–33427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.R.; Wang, H.Y.; Ding, N.; Lu, T.; Huang, Y.C.; Xiao, H.X.; Liu, B.; Li, L. Rapid divergence followed by adaptation to contrasting ecological niches of two closely related columbine species Aquilegia japonica and A. oxysepala. Genome Biol. Evol. 2019, 11, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado-Schiaffino, G.; Henning, F.; Meyer, A. Species-specific differences in adaptive phenotypic plasticity in an ecologically relevant trophic trait: Hypertrophic lips in Midas cichlid fishes. Evolution 2014, 68, 2086–2091. [Google Scholar] [CrossRef] [Green Version]
- Bossdorf, O.; Richards, C.L.; Massimo, P. Epigenetics for ecologists. Ecol. Lett. 2008, 11, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Diez, C.M.; Roessler, K.; Gaut, B.S. Epigenetics and plant genome evolution. Curr. Opin. Plant Biol. 2014, 18, 1–8. [Google Scholar] [CrossRef]
- Verhoeven, K.J.; Vonholdt, B.M.; Sork, V.L. Epigenetics in ecology and evolution: What we know and what we need to know. Mol. Ecol. 2016, 25, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.L.; Oliver, B.; Verhoeven, K.J.F. Understanding natural epigenetic variation. New Phytol. 2010, 187, 562–564. [Google Scholar] [CrossRef]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, B.; Gogol-Döring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Cortijo, S.; Wardenaar, R.; Colomé-Tatché, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.-M.; Wincker, P.; et al. Mapping the epigenetic basis of complex traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef]
- Fisher, V.A.; Wang, L.; Deng, X.; Sarnowski, C.; Cupples, L.A.; Liu, C.T. Do changes in DNA methylation mediate or interact with SNP variation? A pharmacoepigenetic analysis. BMC Genet. 2018, 19, 70. [Google Scholar] [CrossRef]
- Li, L.; Wang, C.; Lu, T.; Lin, S.; Hu, Y.Q. Indirect effect inference and application to GAW20 data. BMC Genet. 2018, 19, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [Green Version]
- Zilberman, D. An evolutionary case for functional gene body methylation in plants and animals. Genome Biol. 2017, 18, 87. [Google Scholar] [CrossRef] [Green Version]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant-Carrying | Reference | Chromosome | Position | Reference Allele | Variant | Annotation | Gene Function |
|---|---|---|---|---|---|---|---|---|
| Aqcoe1G273400 | Asian | American | Chr1 | 18994915 | GAA | GAAA | frameshift | DNA mismatch repair protein MutS2 |
| Aqcoe2G151500 | European | American | Chr2 | 15305837 | A | G | splicing | PIF1-like helicase |
| European | American | 15307442 | A | C | stop gain | |||
| European | American | 15309865 | AATATATAT | AATATATATAT | frameshift | |||
| European | Asian | 15307442 | A | C | stop gain | |||
| European | Asian | 15309865 | AATATATAT | AATATATATAT | frameshift | |||
| A. oxysepala | A. japonica | 15305837 | A | G | splicing | |||
| A. oxysepala | A. japonica | 15309267 | AT | A | frameshift | |||
| Aqcoe2G177700 | European | American | Chr2 | 21794397 | TATGCACCAAAGGTATCACGATGC | TATGC | frameshift | PIF1-like helicase |
| European | American | 21794979 | TT | TTGT | frameshift | |||
| European | Asian | 21794397 | TATGCACCAAAGGTATCACGATGC | TATGC | frameshift | |||
| A. oxysepala | A. japonica | 21795089 | CA | C | frameshift | |||
| Aqcoe6G208600 | European | American | Chr6 | 15364081 | A | ATCTCTTCG | frameshift | PIF1-like helicase |
| European | Asian | 15364081 | A | ATCTCTTCG | frameshift | |||
| A. japonica | A. oxysepala | 15364330 | TAA | TA | frameshift | |||
| Aqcoe6G253800 | European | American | Chr6 | 22789898 | C | T | stop gain | DNA helicase |
| European | American | 22790012 | G | A | splicing | |||
| European | Asian | 22789898 | C | T | stop gain | |||
| A. japonica | A. oxysepala | 22790012 | G | A | splicing | |||
| Aqcoe2G276600 | Asian | American | Chr2 | 33314422 | AGGGGG | AGGGGGG | frameshift | DNA mismatch repair protein Msh6 |
| Aqcoe6G160300 | A. japonica | A. oxysepala | Chr6 | 9414625 | G | A | stop gain | TPX2 |
| Aqcoe7G062500 | A. oxysepala | A. japonica | Chr7 | 3789055 | G | A | stop gain | cell cycle-regulated microtubule-associated protein |
| Type of Selection | Differential Methylation | Jap-Oxy * | Jap-Ame | Jap-Eur | Oxy-Ame | Oxy-Eur | Ame-Eur |
|---|---|---|---|---|---|---|---|
| DMG | 7.2% | 7.3% | 11.9% | 6.7% | 8.4% | 8.9% | |
| Positive selection | non-DMG | 4.4% | 5.0% | 5.6% | 4.7% | 5.4% | 5.4% |
| p value | 0.11 | 7.3 × 10−2 | 3.9 × 10−5 | 6.7 × 10−2 | 1.8 × 10−2 | 2.8 × 10−4 | |
| DMG | 3.1% | 1.8% | 2.4% | 2.3% | 2.0% | 1.9% | |
| Purifying selection | non-DMG | 4.3% | 4.3% | 4.7% | 4.9% | 5.1% | 4.0% |
| p value | 0.53 | 3.2 × 10−2 | 9.1 × 10−2 | 1.3 × 10−2 | 8.4 × 10−3 | 1.0 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Lu, T.; Li, M.; Ding, N.; Lan, L.; Gao, X.; Xiong, A.; Zhang, J.; Li, L. Genetic and Epigenetic Signatures Associated with the Divergence of Aquilegia Species. Genes 2022, 13, 793. https://doi.org/10.3390/genes13050793
Wang Z, Lu T, Li M, Ding N, Lan L, Gao X, Xiong A, Zhang J, Li L. Genetic and Epigenetic Signatures Associated with the Divergence of Aquilegia Species. Genes. 2022; 13(5):793. https://doi.org/10.3390/genes13050793
Chicago/Turabian StyleWang, Zhenhui, Tianyuan Lu, Mingrui Li, Ning Ding, Lizhen Lan, Xiang Gao, Aisheng Xiong, Jian Zhang, and Linfeng Li. 2022. "Genetic and Epigenetic Signatures Associated with the Divergence of Aquilegia Species" Genes 13, no. 5: 793. https://doi.org/10.3390/genes13050793