
Genome-Wide Characterization and Expression Analysis of GATA Transcription Factors in Response to Methyl Jasmonate in Salvia miltiorrhiza

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification and Sequence Analysis of S. miltiorrhiza GATA Genes
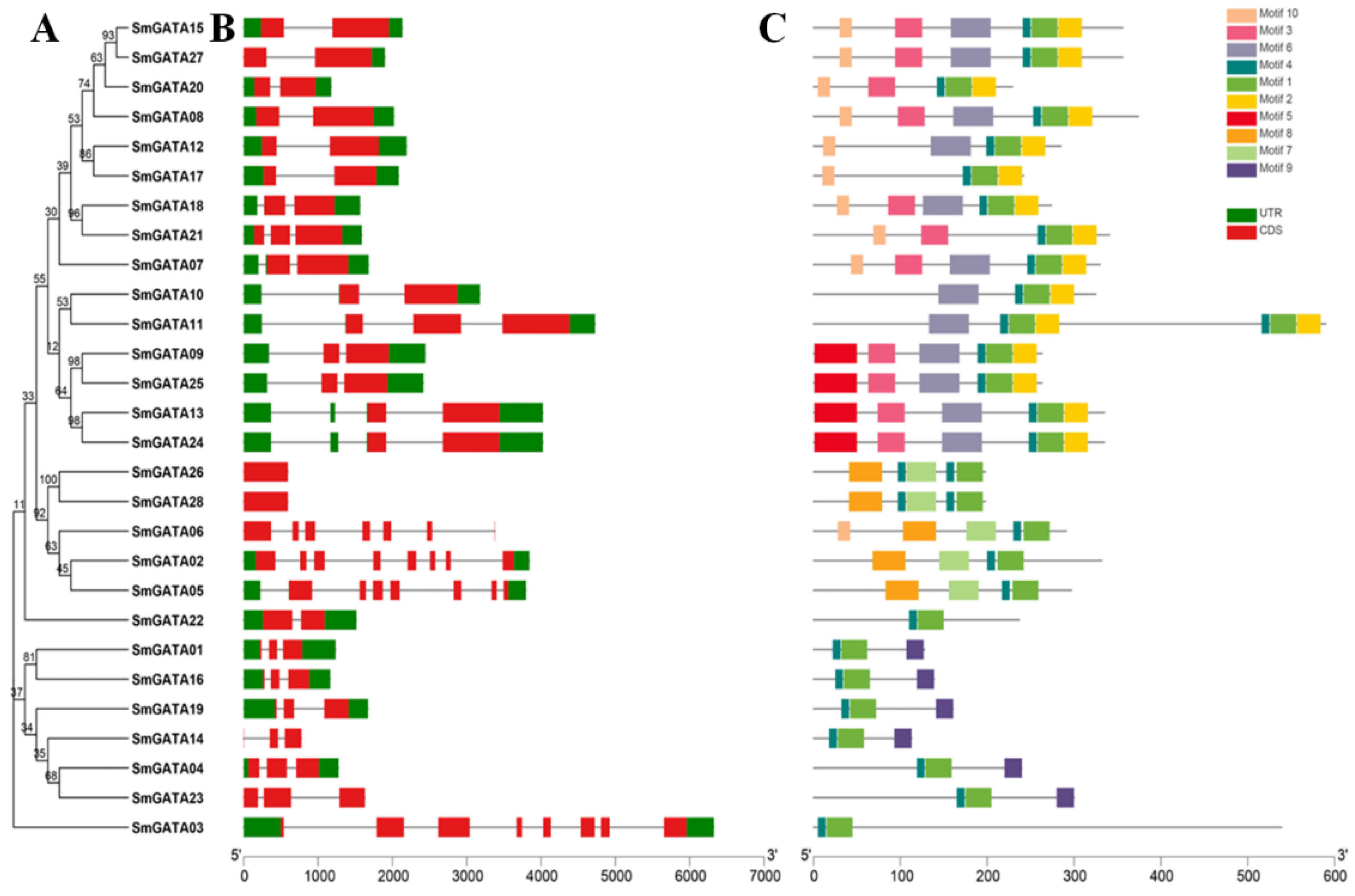
2.2. Phylogenetic and Conserved Domains Analysis
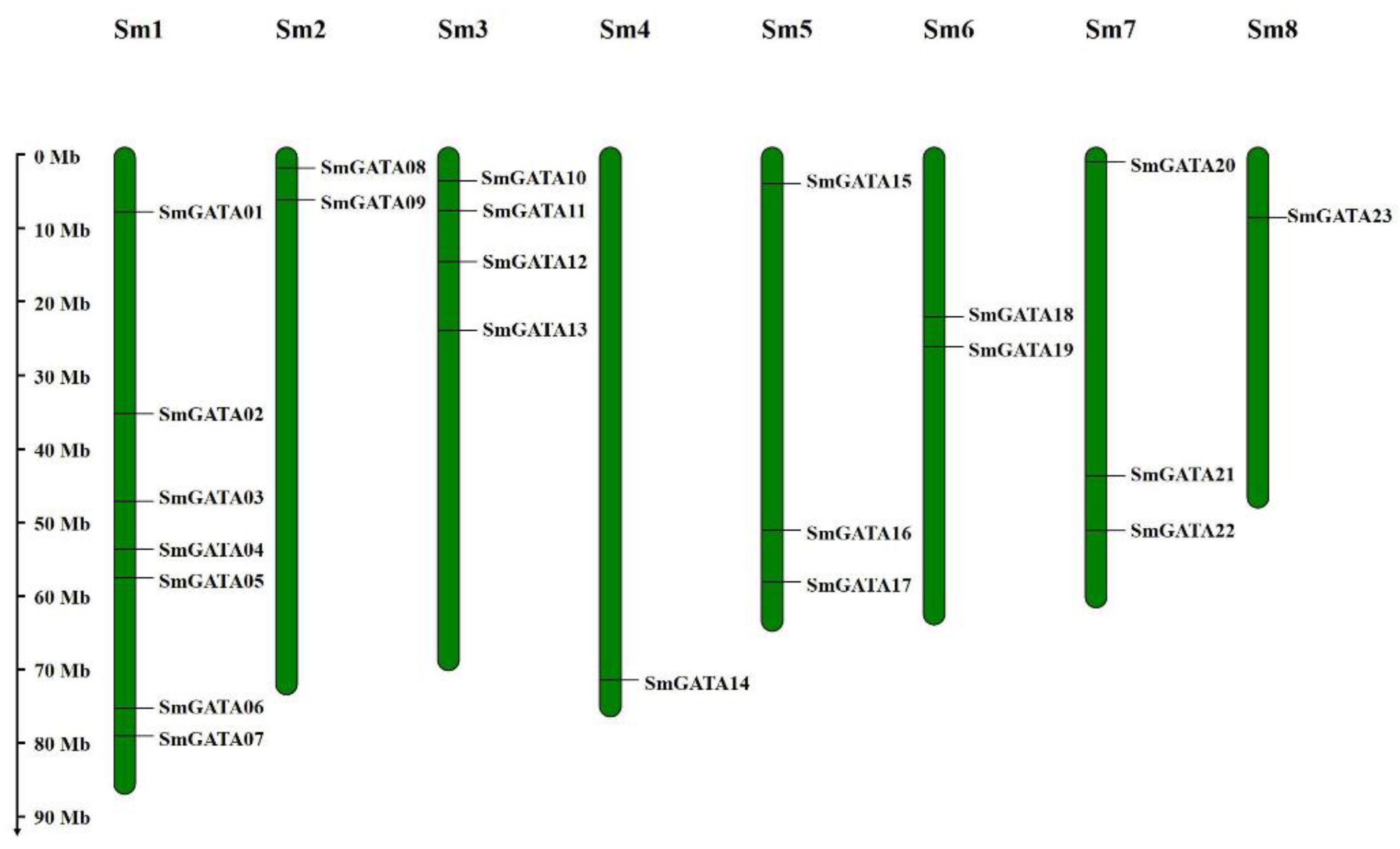
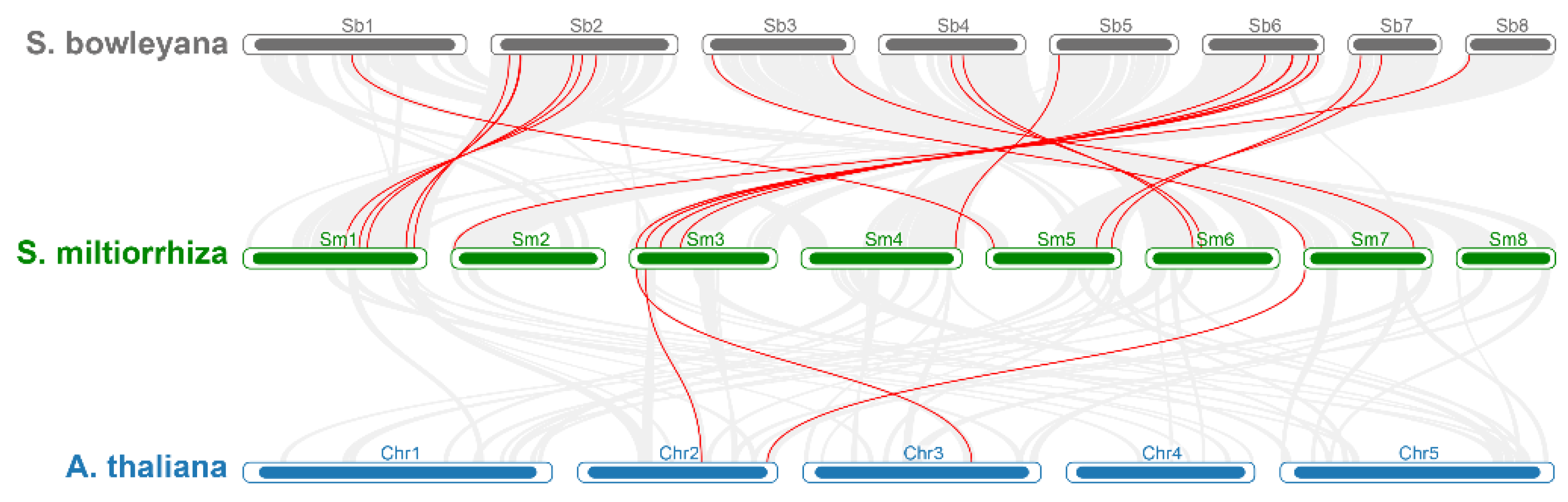
2.3. Chromosome Localizations and Genomic Synteny of SmGATA Genes
2.4. Gene Structure and Conserved Motif Analysis of SmGATA Genes
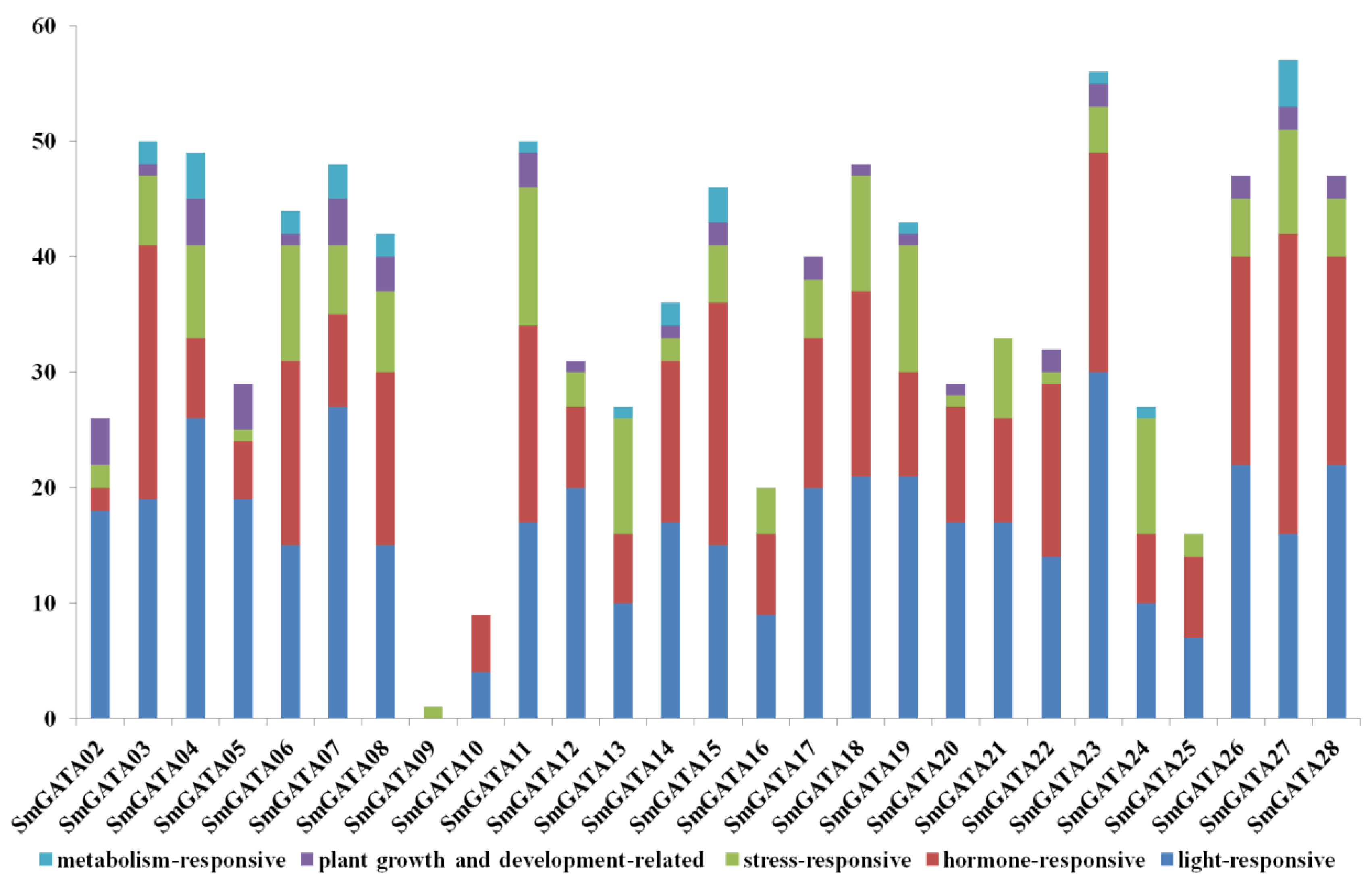
2.5. Cis-Acting Regulatory Element Analysis of SmGATA Genes
2.6. Expression Profiles of SmGATA Genes Based on Transcriptome Datasets
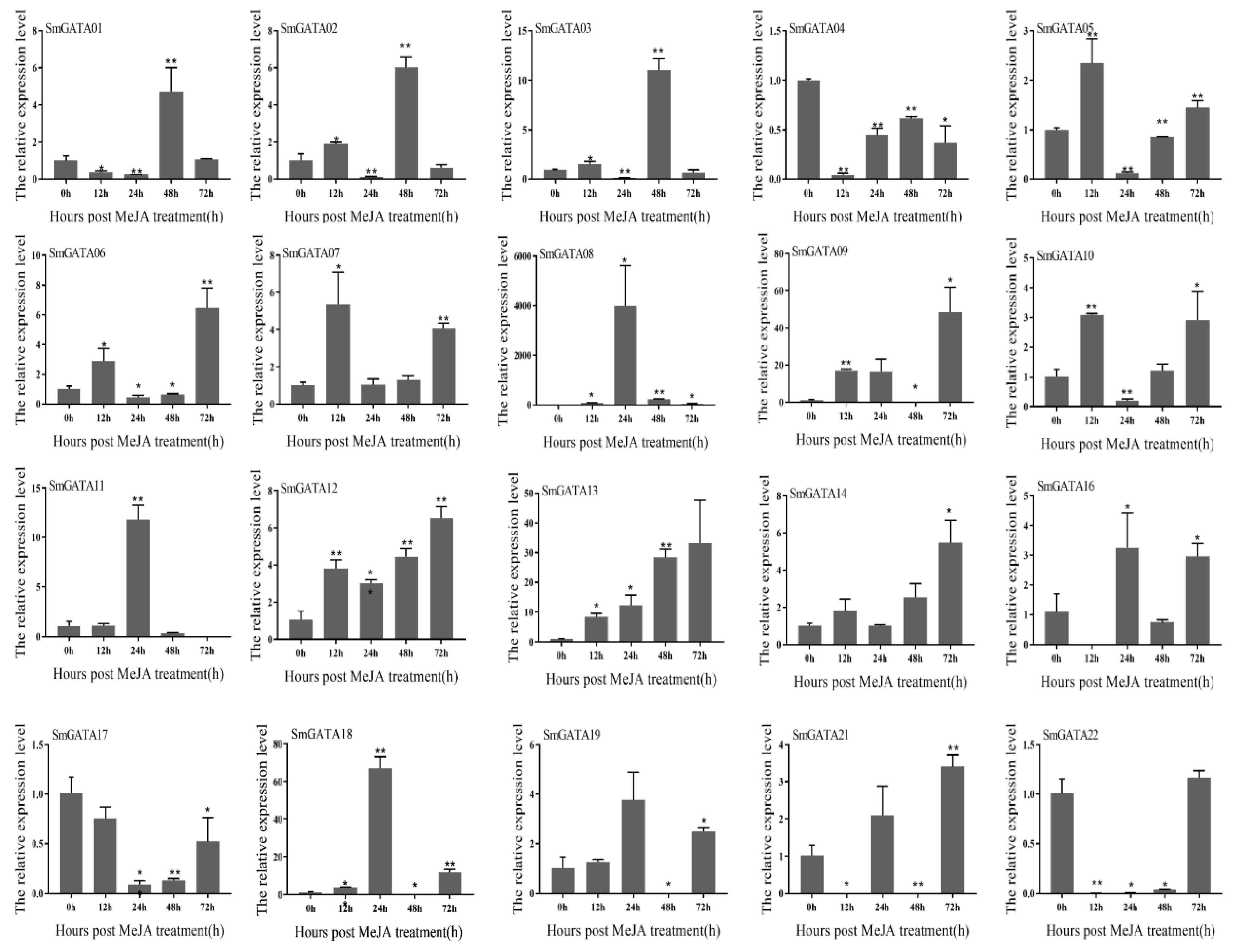
2.7. Expression Analysis of SmGATAs under MeJA Treatment
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Identification and Sequence Analysis of SmGATA Genes
5.2. Phylogenetic Analysis of GATA Genes
5.3. Gene Structure and Conserved Motifs Analysis
5.4. Analysis of Cis-Acting Elements in the Promoters of SmGATA Genes
5.5. Chromosomal Localization and Collinearity Analysis
5.6. Expression Profiles of SmGATAs in S. miltiorrhiza
5.7. Plant Materials and MeJA Treatments
5.8. Quantitative Real-Time PCR (qRT-PCR) Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lowry, J.A.; Atchley, W.R. Molecular evolution of the GATA family of transcription factors: Conservation within the DNA-binding domain. J. Mol. Evol. 1999, 50, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Daniel-Vedele, F.; Caboche, M. A tobacco cDNA clone encoding a GATA-1 zinc finger protein homologous to regulators of nitrogen metabolism in fungi. Mol. Gen. Genet. 1993, 240, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.C.; Muro-Pastor, M.I.; Florencio, F.J. The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol. 2004, 134, 1718–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behringer, C.; Schwechheimer, C. B-GATA transcription factors-insights into their structure, regulation, and role in plant development. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwechheimer, C.; Schröder, P.M.; Blaby-Haas, C.E. Plant GATA Factors: Their Biology, Phylogeny, and Phylogenomics. Annu. Rev. Plant Biol. 2022. epub ahead of print. [Google Scholar] [CrossRef]
- Richter, R.; Behringer, C.; Zourelidou, M.; Schwechheimer, C. Convergence of auxin and gibberellins signaling on the regulation of the GATA transcription factors GNC and GNL in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2013, 110, 13192–13197. [Google Scholar] [CrossRef] [Green Version]
- Richter, R.; Bastakis, E.; Schwechheimer, C. Cross-repressive interactions between SOC1 and the GATAs GNC and GNL/CGA1 in the control of greening, cold tolerance, and flowering time in Arabidopsis. Plant Physiol. 2013, 162, 1992–2004. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hou, Y.; Hao, Q.; Chen, H.; Chen, L.; Yuan, S.; Shan, Z.; Zhang, X.; Yang, Z.; Qiu, D.; et al. Genome-Wide Survey of the Soybean GATA Transcription Factor Gene Family and Expression Analysis under Low Nitrogen Stress. PLoS ONE 2015, 10, e0125174. [Google Scholar]
- An, Y.; Zhou, Y.; Han, X.; Shen, C.; Wang, S.; Liu, C.; Yin, W.; Xia, X. The GATA transcription factor GNC plays an important role in photosynthesis and growth in poplar. J. Exp. Bot. 2019, 71, 1969–1984. [Google Scholar] [CrossRef]
- Richter, R.; Behringer, C.; Müller, I.K.; Schwechheimer, C. The GATA-type transcription factors GNC and GNL/CGA1 repress gibberellin signaling downstream from DELLA proteins and phytochrome-interacting factors. Genes Dev. 2010, 24, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Mangi, K.; Hong, X.; Jongsun, P. Genome-wide comparative analyses of GATA transcription factors among 19 Arabidopsis ecotype genomes: Intraspecific characteristics of GATA transcription factors. PLoS ONE 2021, 16, e0252181. [Google Scholar]
- Chiang, Y.-H.; Zubo, Y.O.; Tapken, W.; Kim, H.J.; Lavanway, A.M.; Howard, L.; Pilon, M.; Kieber, J.J.; Schaller, G.E. Functional characterization of the GATA transcription factors GNC and CGA1 reveals their key role in chloroplast development, growth, and division in Arabidopsis. Plant Physiol. 2012, 160, 332–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.-M.; Lin, W.-H.; Zhu, S.; Zhu, J.-Y.; Sun, Y.; Cheng, M.; Hao, Y.; Oh, E.; Tian, M.; Liu, L.; et al. Integration of light- and brassinosteroidsignaling pathways by a GATA transcription factor in Arabidopsis. Dev. Cell 2010, 19, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Yung, V.; Chiba, T.; Feldman, L.J. Longitudinal patterning in roots: A GATA2–auxin interaction underlies and maintains the root transition domain. Planta 2018, 247, 831–843. [Google Scholar] [CrossRef]
- Ravindran, P.; Verma, V.; Stamm, P.; Kumar, P.P. A Novel RGL2–DOF6 Complex Contributes to Primary Seed Dormancy in Arabidopsis thaliana by Regulating a GATA Transcription Factor. Mol. Plant 2017, 10, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Bi, Y.-M.; Rothstein, S.J. Overexpression of OsGATA12 regulates chlorophyll content, delays plant senescence and improves rice yield under high density planting. Plant Mol. Biol. 2017, 94, 215–227. [Google Scholar] [CrossRef]
- He, P.; Wang, X.; Zhang, X.; Jiang, Y.; Tian, W.; Zhang, X.; Li, Y.; Sun, Y.; Xie, J.; Ni, J.; et al. Short and narrow flag leaf1, a GATA zinc finger domain-containing protein, regulates flag leaf size in rice (Oryza sativa). BMC Plant Biol. 2018, 18, 273. [Google Scholar] [CrossRef]
- Hudson, D.; Guevara, D.R.; Hand, A.J.; Xu, Z.; Hao, L.; Chen, X.; Zhu, T.; Bi, Y.-M.; Rothstein, S.J. Rice cytokinin GATA transcription Factor1 regulates chloroplast development and plant architecture. Plant Physiol. 2013, 162, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Flora of China Editorial Committee of Chinese Academy of Sciences. Flora of China; Science Press: Beijing, China, 1996; Volume 1, p. 252. [Google Scholar]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2020; Volume 1. [Google Scholar]
- Xu, H.; Song, J.; Luo, H.; Zhang, Y.; Li, Q.; Zhu, Y.; Xu, J.; Li, Y.; Song, C.; Wang, B.; et al. Analysis of the genome sequence of the medicinal plant Salviamiltior-rhiza. Mol. Plant 2016, 9, 949–952. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Zhu, Y.; Song, J.; Xu, L.; Sun, C.; Zhang, X.; Xu, Y.; He, L.; Sun, W.; Xu, H.; et al. Transcriptional data mining of Salvia miltiorrhiza inresponse to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation. Physiol. Plant. 2014, 152, 241–255. [Google Scholar] [CrossRef]
- Shi, M.; Zhou, W.; Zhang, J.; Huang, S.; Wang, H.; Kai, G. Methyl jasmonate induction of tanshinone biosynthesis in Salvia miltiorrhiza hairy roots is mediated by JASMONATE ZIM-DOMAIN repressor proteins. Sci. Rep. 2016, 6, 20919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Yang, D.; Liang, Z. Diverse responses to methyl jasmonate in hairy roots of two Salvia species. J. Zhejiang Sci-Tech. Univ. Nat. Sci. 2017, 37, 712–719. [Google Scholar]
- Xing, B.; Yang, D.; Liu, L.; Han, R.; Sun, Y.; Liang, Z. Phenolic acid production is more effectively enhanced than tanshinone production by methyl jasmonate in Salvia miltiorrhiza hairy roots. Plant Cell Tiss Organ Cult. 2018, 134, 119–129. [Google Scholar] [CrossRef]
- Hou, Z.; Li, Y.; Su, F.; Chen, J.; Zhang, X.; Xu, L.; Yang, D.; Liang, Z. Application of 1 H-NMR combined with qRT-PCR technology in the exploration of rosmarinic acid biosynthesis in hair roots of Salvia miltiorrhiza Bunge and Salvia castanea f. tomentosa Stib. Planta 2021, 2, 256. [Google Scholar] [CrossRef]
- Teakle, G.R.; Manfield, I.W.; Graham, J.F.; Gilmartin, P.M. Arabidopsis thaliana GATA factors: Organisation, expression and DNA-binding characteristics. Plant Mol. Biol. 2002, 50, 43–57. [Google Scholar] [CrossRef]
- Jeong, M.J.; Shih, M.C. Interaction of a GATA factor with cis-acting elements involved in light regulation of nuclear genes encoding chloroplast glyceraldehyde-3-phosphate dehydrogenase in Arabidopsis. Biochem. Biophys. Res. Commun. 2003, 300, 555–562. [Google Scholar] [CrossRef]
- Naito, T.; Kiba, T.; Koizumi, N.; Yamashino, T.; Mizuno, T. Characterization of a Unique GATA Family Gene That Responds to Both Light and Cytokinin in Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 2007, 71, 1557–1560. [Google Scholar] [CrossRef] [Green Version]
- Ranftl, Q.L.; Bastakis, E.; Klermund, C.; Schwechheimer, C. LLM-Domain Containing B-GATA Factors Control Different Aspects of Cytokinin-Regulated Development in Arabidopsis thaliana. Plant Physiol. 2016, 170, 2295–2311. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shao, H.; Li, K.; Zhang, D.; Fan, S.; Li, Y.; Han, M. Genome-wide identification, evolution, and expression analysis of GATA transcription factors in apple (Malus × domestica Borkh). Gene 2017, 627, 460–472. [Google Scholar] [CrossRef]
- Ni, P.; Ji, X.; Guo, D. Genome-wide indentification, characterization, and expression analysis of GDSL-type esterases/lipases gene family in relation to grape berry ripening. Sci. Hortic. 2020, 264, 109162. [Google Scholar] [CrossRef]
- Zhu, W.; Guo, Y.; Chen, Y.; Wu, D.; Jiang, L. Genome-wide identification, phylogenetic and expression pattern analysis of GATA family genes in Brassica napus. BMC Plant Biol. 2020, 20, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Nishii, A.; Takemura, M.; Fujita, H.; Shikata, M.; Yokota, A.; Kohchi, T. Characterization of a novel gene encoding a putative single zinc-finger protein, ZIM, expressed during the reproductive phase in Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 2000, 64, 1402–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikhali, J.; de Dios Barajas-Lopéz, J.; Ötvös, K.; Kremnev, D.; Garcia, A.S.; Srivastava, V.; Wingsle, G.; Bako, L.; Strand, Å. The CRYPTOCHROME1-Dependent Response to Excess Light Is Mediated through the Transcriptional Activators ZINC FINGER PROTEIN EXPRESSED IN INFLORESCENCE MERISTEM LIKE1 and ZML2 in Arabidopsis. Plant Cell 2012, 24, 3009–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholme, B.; Grunewald, W.; Bateman, A.; Kohchi, T.; Gheysen, G. The tify family previously known as ZIM. Trends Plant Sci. 2007, 12, 239–244. [Google Scholar] [CrossRef]
- Baskind, H.A.; Na, L.; Ma, Q.; Patel, M.P.; Geenen, D.L.; Wang, Q. Functional conservation of Asxl2, a murine homolog for the drosophila enhancer of trithorax and polycomb group gene Asx. PLoS ONE 2009, 4, e4750. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.L.; Lee, I.; Bloyer, S.; Bozza, S.; Chevalier, J.; Dahl, A.; Bodner, C.; Helgason, C.D.; Hess, J.L.; Humphries, R.K.; et al. Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev. Biol. 2010, 337, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Iyer, L.M. The HARE-HTH and associated domains: Novel modules in the coordination of epigenetic DNA and protein modifications. Cell Cycle 2012, 11, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Kiryushkin, A.S.; Ilina, E.L.; Puchkova, V.A.; Guseva, E.D.; Pawlowski, K.; Demchenko, K.N. Lateral root initiation in the parental root meristem of cucurbits: Old players in a new position. Front. Plant Sci. 2019, 10, 365. [Google Scholar] [CrossRef]
- Seo, D.H.; Seomun, S.; Choi, Y.D.; Jang, G. Root development and stress tolerance in rice: The key to improving stress tolerance without yield penalties. Int. J. Mol. Sci. 2020, 21, 1807. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Ma, Y.; Wu, S.; Zheng, X.; Kang, H.; Sang, J.; Xu, X.; Hao, L.; Li, Z.; Gong, Z.; et al. Genome Warehouse: A Public Repository Housing Genome-scale Data. Genom. Proteom. Bioinform. 2021, 19, 584–589. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in Homology Search: HMMER3 and Convergent Evolution of Coiled-Coil Regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, 597–603. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.; Shen, H. A New Method for Predicting the Subcellular Localization of Eukaryotic Proteins with Both Single and Multiple Sites: Euk-mPLoc 2.0. PLoS ONE 2010, 5, e9931. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Jin, J.; Guo, A.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvement in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Liu, T.; Wang, B.; Li, H. Genome-Wide Characterization and Expression Analysis of GATA Transcription Factors in Response to Methyl Jasmonate in Salvia miltiorrhiza. Genes 2022, 13, 822. https://doi.org/10.3390/genes13050822
Li H, Liu T, Wang B, Li H. Genome-Wide Characterization and Expression Analysis of GATA Transcription Factors in Response to Methyl Jasmonate in Salvia miltiorrhiza. Genes. 2022; 13(5):822. https://doi.org/10.3390/genes13050822
Chicago/Turabian StyleLi, Haiyan, Tianrang Liu, Biao Wang, and Hongbo Li. 2022. "Genome-Wide Characterization and Expression Analysis of GATA Transcription Factors in Response to Methyl Jasmonate in Salvia miltiorrhiza" Genes 13, no. 5: 822. https://doi.org/10.3390/genes13050822
APA StyleLi, H., Liu, T., Wang, B., & Li, H. (2022). Genome-Wide Characterization and Expression Analysis of GATA Transcription Factors in Response to Methyl Jasmonate in Salvia miltiorrhiza. Genes, 13(5), 822. https://doi.org/10.3390/genes13050822