
Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods


Abstract
:1. Introduction
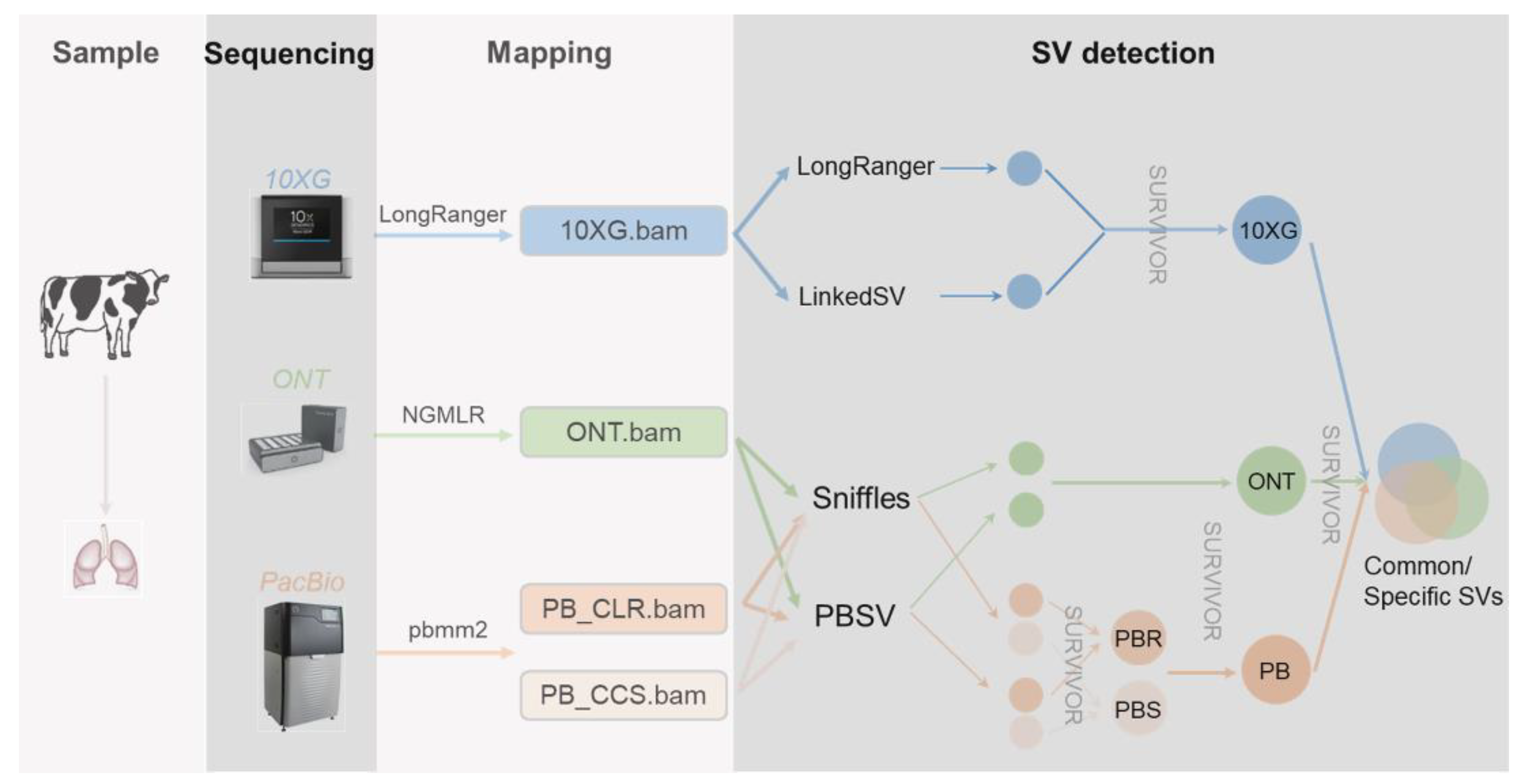
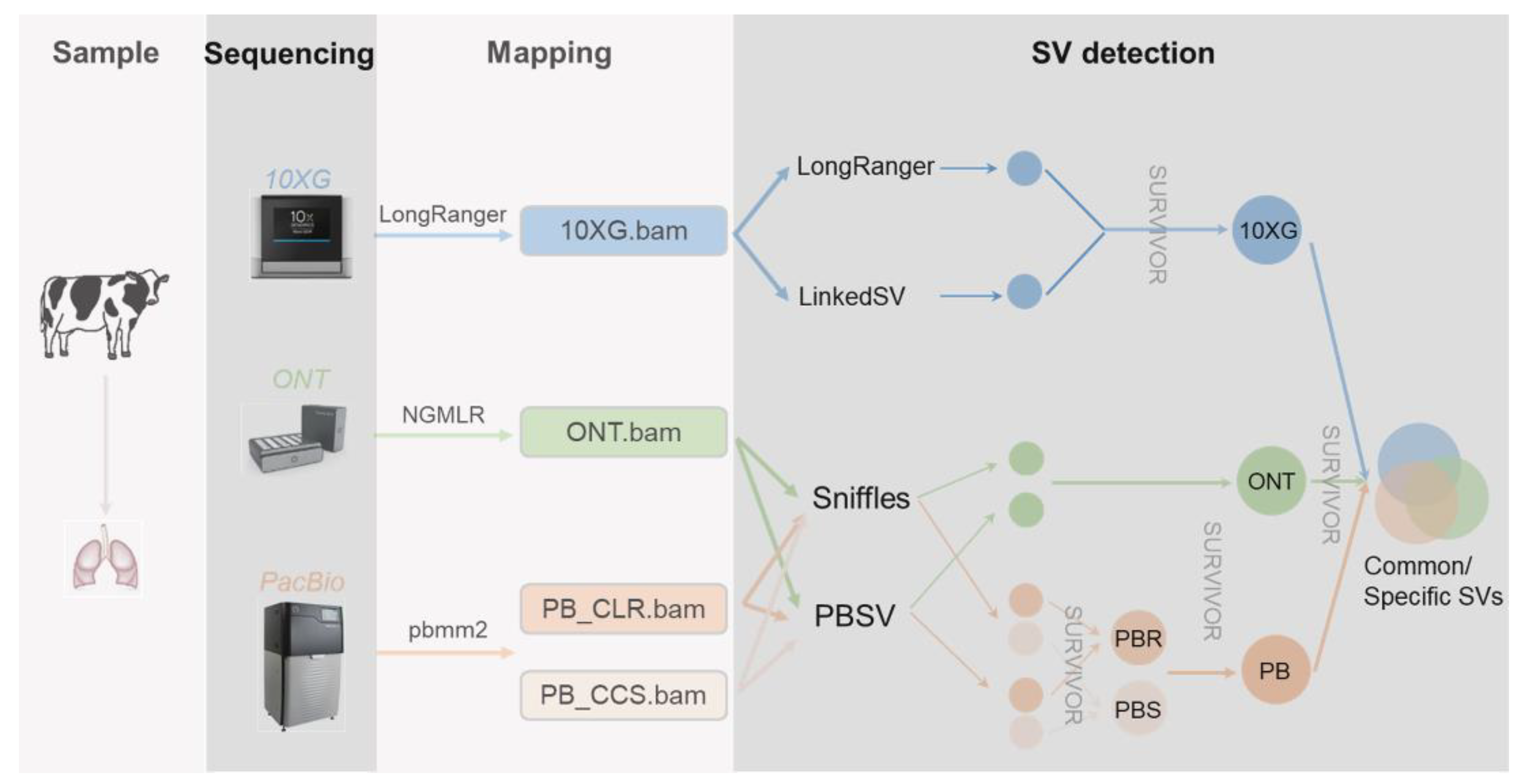
2. Materials and Methods
3. Results
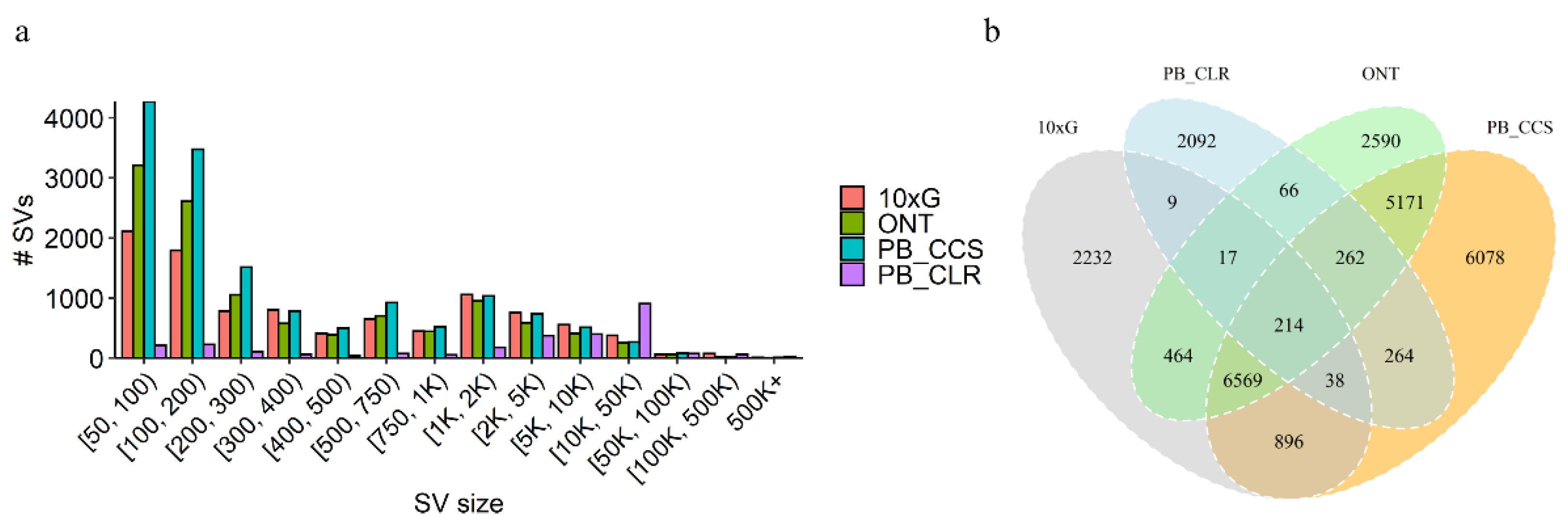
3.1. SV Inference
3.2. SV Overlap
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaisson, M.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; GTEx Consortium; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaoğlanoğlu, F.; Ricketts, C.; Ebren, E.; Rasekh, M.E.; Hajirasouliha, I.; Alkan, C. VALOR2: Characterization of large-scale structural variants using linked-reads. Genome Biol. 2020, 21, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A.; et al. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spies, N.; Weng, Z.; Bishara, A.; McDaniel, J.; Catoe, D.; Zook, J.M.; Salit, M.; West, R.B.; Batzoglou, S.; Sidow, A. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 2017, 14, 915–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretu Stancu, M.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Espejo Valle-Inclan, J.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Rhie, A.; Walenz, B.P.; Dilthey, A.T.; Bickhart, D.M.; Kingan, S.B.; Hiendleder, S.; Williams, J.L.; Smith, T.; Phillippy, A.M. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 2018, 36, 174–1182. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Serra Mari, R.; et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372, eabf7117. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Li, Y.; Liu, X.; Wang, Y.; Ping, J.; Lu, Y.; Zhou, G. Characterization of structural variation in Tibetans reveals new evidence of high-altitude adaptation and introgression. Genome Biol. 2021, 22, 159. [Google Scholar] [CrossRef]
- Low, W.Y.; Tearle, R.; Liu, R.; Koren, S.; Rhie, A.; Bickhart, D.M.; Rosen, B.D.; Kronenberg, Z.N.; Kingan, S.B.; Tseng, E.; et al. Haplotype-resolved genomes provide insights into structural variation and gene content in Angus and Brahman cattle. Nat. Commun. 2020, 11, 2071. [Google Scholar] [CrossRef]
- Couldrey, C.; Keehan, M.; Johnson, T.; Tiplady, K.; Winkelman, A.; Littlejohn, M.D.; Scott, A.; Kemper, K.E.; Hayes, B.; Davis, S.R.; et al. Detection and assessment of copy number variation using PacBio long-read and Illumina sequencing in New Zealand dairy cattle. J. Dairy Sci. 2017, 100, 5472–5478. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.J.; Ross, E.M.; Nguyen, L.T.; Lyons, R.E.; Moore, S.S.; Hayes, B.J. Characterization of the poll allele in Brahman cattle using long-read Oxford Nanopore sequencing. J. Anim. Sci. 2020, 98, skaa127. [Google Scholar] [CrossRef]
- Ananthasayanam, S.; Kothandaraman, H.; Nayee, N.; Saha, S.; Baghel, D.S.; Gopalakrishnan, K.; Peddamma, S.; Singh, R.B.; Schatz, M. First near complete haplotype phased genome assembly of River buffalo (Bubalus bubalis). bioRxiv 2020, 618785. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Li, S.T.; Yao, W.Y.; Xie, C.D.; Chen, Z.; Zeng, Z.J.; Wang, D.; Xu, K.; Shen, Z.J.; Mu, Y.; et al. The Meishan pig genome reveals structural variation-mediated gene expression and phenotypic divergence underlying Asian pig domestication. Mol. Ecol. Resour. 2021, 21, 2077–2092. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Jiang, J.; He, J.; Liu, H.; Han, L.; Gong, Y.; Li, B.; Yu, Z.; Tang, S.; Zhang, Y.; et al. Long-read assembly of the Chinese indigenous Ningxiang pig genome and identification of genetic variations in fat metabolism among different breeds. Mol. Ecol. Resour. 2022, 22, 1508–1520. [Google Scholar] [CrossRef]
- Li, R.; Gong, M.; Zhang, X.; Wang, F.; Liu, Z.; Zhang, L.; Xu, M.; Zhang, Y.; Dai, X.; Zhang, Z.; et al. The first sheep graph-based pan-genome 1 reveals the spectrum of structural variations and their effects on tail phenotypes. bioRxiv 2021, 472709. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aganezov, S.; Goodwin, S.; Sherman, R.M.; Sedlazeck, F.J.; Arun, G.; Bhatia, S.; Lee, I.; Kirsche, M.; Wappel, R.; Kramer, M.; et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res. 2020, 30, 1258–1273. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Kao, C.; Gonzalez, M.V.; Mafra, F.A.; Pellegrino da Silva, R.; Li, M.; Wenzel, S.S.; Wimmer, K.; Hakonarson, H.; Wang, K. LinkedSV for detection of mosaic structural variants from linked-read exome and genome sequencing data. Nat. Commun. 2019, 10, 5585. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. [Google Scholar] [CrossRef] [Green Version]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- English, A.C.; Salerno, W.J.; Hampton, O.A.; Gonzaga-Jauregui, C.; Ambreth, S.; Ritter, D.I.; Beck, C.R.; Davis, C.F.; Dahdouli, M.; Ma, S.; et al. Assessing structural variation in a personal genome-towards a human reference diploid genome. BMC Genom. 2015, 16, 286. [Google Scholar] [CrossRef] [Green Version]
- Leonard, A.S.; Crysnanto, D.; Fang, Z.H.; Heaton, M.P.; Ley, B.L.V.; Herrera, C.; Bollwein, H.; Bickhart, D.M.; Kuhn, K.L.; Smith, T.P.L.; et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. bioRxiv 2021, 466900. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Platform | 10 × G | PromethION | PacBio CLR | PacBio CCS |
|---|---|---|---|---|
| Number of reads | 1,577,259,728 | 1,618,623 | 11,178,388 | 2,875,796 |
| Mapped reads | 1,532,221,733 | 1,488,641 | 11,178,388 | 2,875,796 |
| Mapping rate (%) | 97.14 | 91.97 | 100 | 100 |
| Depth | 55× | 11× | 40× | 6× |
| Read min length | 19 | 70 | 53 | 74 |
| Read max length | 150 | 248,333 | 369,285 | 47,915 |
| Read mean length | 133.94 | 28,191.59 | 25,259.03 | 8763.78 |
| Platform | Method | DEL | DUP | Total |
|---|---|---|---|---|
| 10 × G | LongRanger | 8242 | 73 | 8315 |
| LinkedSV | 6415 | 38 | 6453 | |
| Merge | 10,325 | 114 | 10,439 | |
| ONT | PBSV | 26,397 | 2888 | 29,285 |
| Sniffles | 3497 | 168 | 3665 | |
| Merge | 13,472 | 1881 | 15,353 | |
| PB_CLR | PBSV | 885 | 169 | 1054 |
| Sniffles | 1340 | 1238 | 2578 | |
| Merge | 1800 | 1162 | 2962 | |
| PB_CCS | PBSV | 23,353 | 6569 | 29,922 |
| Sniffles | 190 | 99 | 289 | |
| Merge | 15,601 | 3891 | 19,492 | |
| Merge | SURVIVOR | 16,289 | 4875 | 21,164 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Ma, L.; Liu, G.E. Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods. Genes 2022, 13, 828. https://doi.org/10.3390/genes13050828
Gao Y, Ma L, Liu GE. Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods. Genes. 2022; 13(5):828. https://doi.org/10.3390/genes13050828
Chicago/Turabian StyleGao, Yahui, Li Ma, and George E. Liu. 2022. "Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods" Genes 13, no. 5: 828. https://doi.org/10.3390/genes13050828
APA StyleGao, Y., Ma, L., & Liu, G. E. (2022). Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods. Genes, 13(5), 828. https://doi.org/10.3390/genes13050828