
Correlation of NTRK1 Downregulation with Low Levels of Tumor-Infiltrating Immune Cells and Poor Prognosis of Prostate Cancer Revealed by Gene Network Analysis

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data Extraction
2.2. Data Preprocessing and Screening of DEGs
2.3. Pathway and Functional Enrichment Analysis
2.4. PPI Network Construction and HUBs Selection
2.5. Overall Survival (OS) Analysis of HUBs
2.6. Validation of Prognostic HUBs Using cBioPortal and UALCAN Databases
2.7. Tumor Infiltration Analysis
3. Results
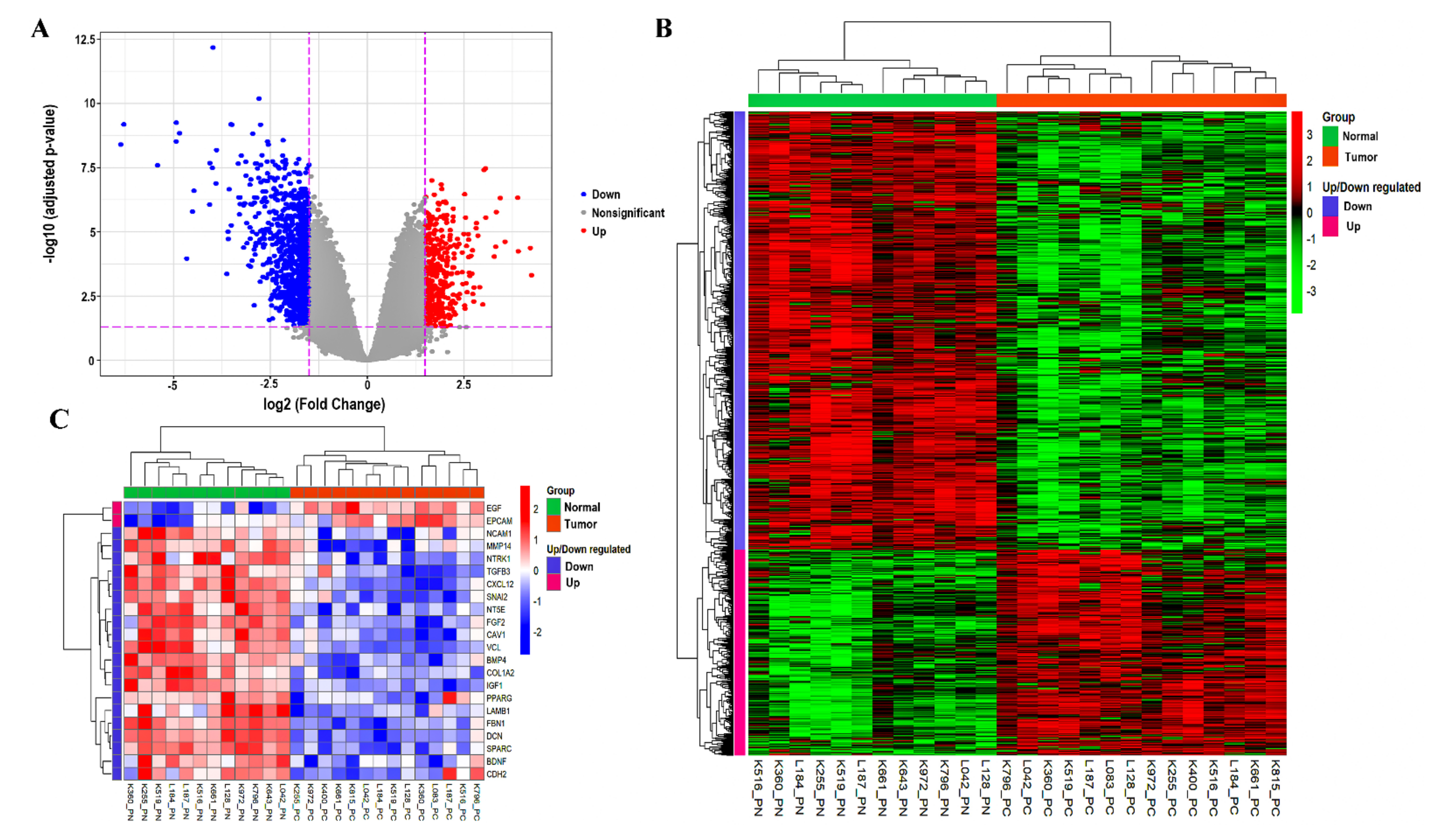
3.1. Data Preprocessing and Identification of DEGs
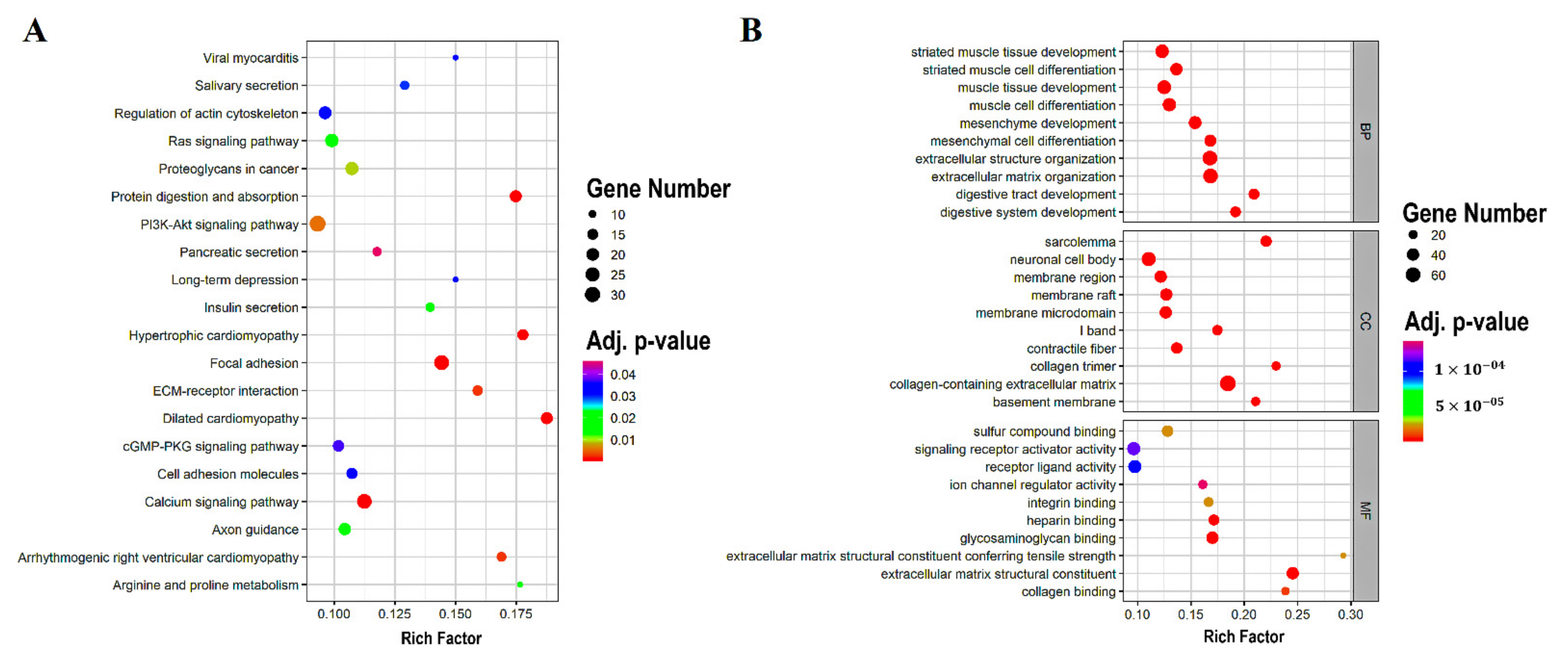
3.2. DEGs Enrichment Analysis
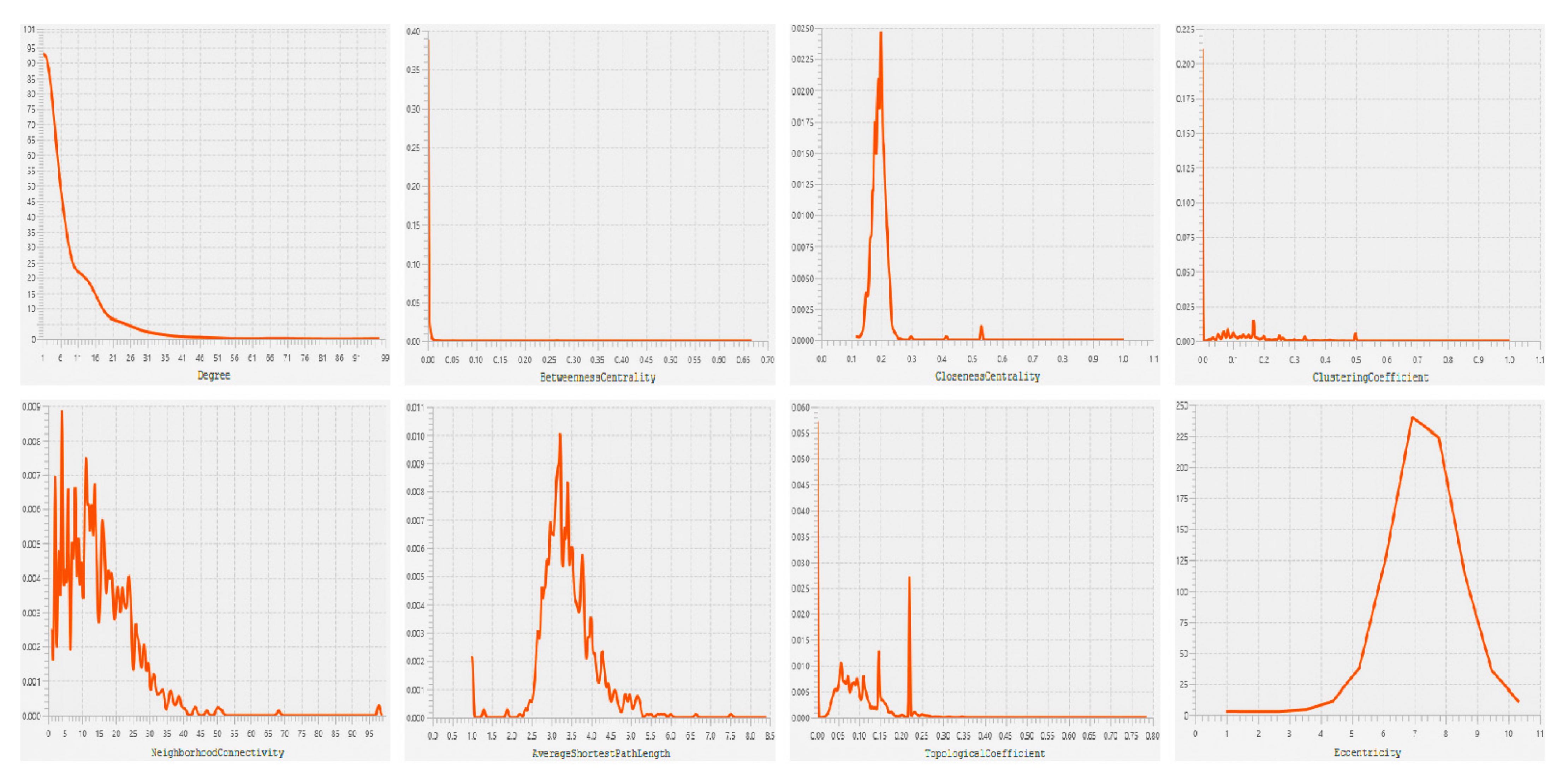
3.3. PPI Network Construction and HUBs Selection
3.4. OS Analysis of HUBs
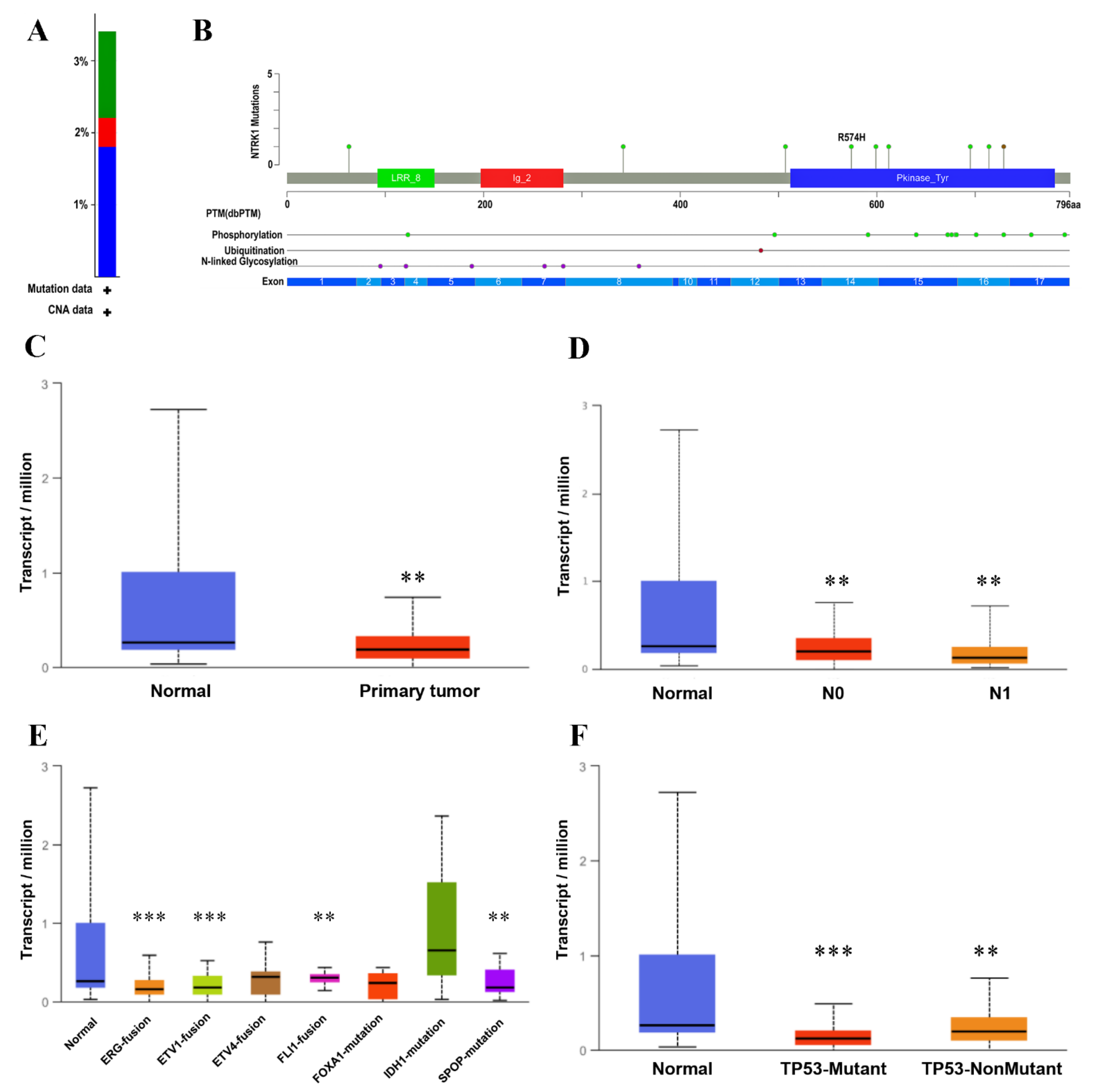
3.5. Validation of Prognostic HUBs Using cBioPortal and UALCAN Databases
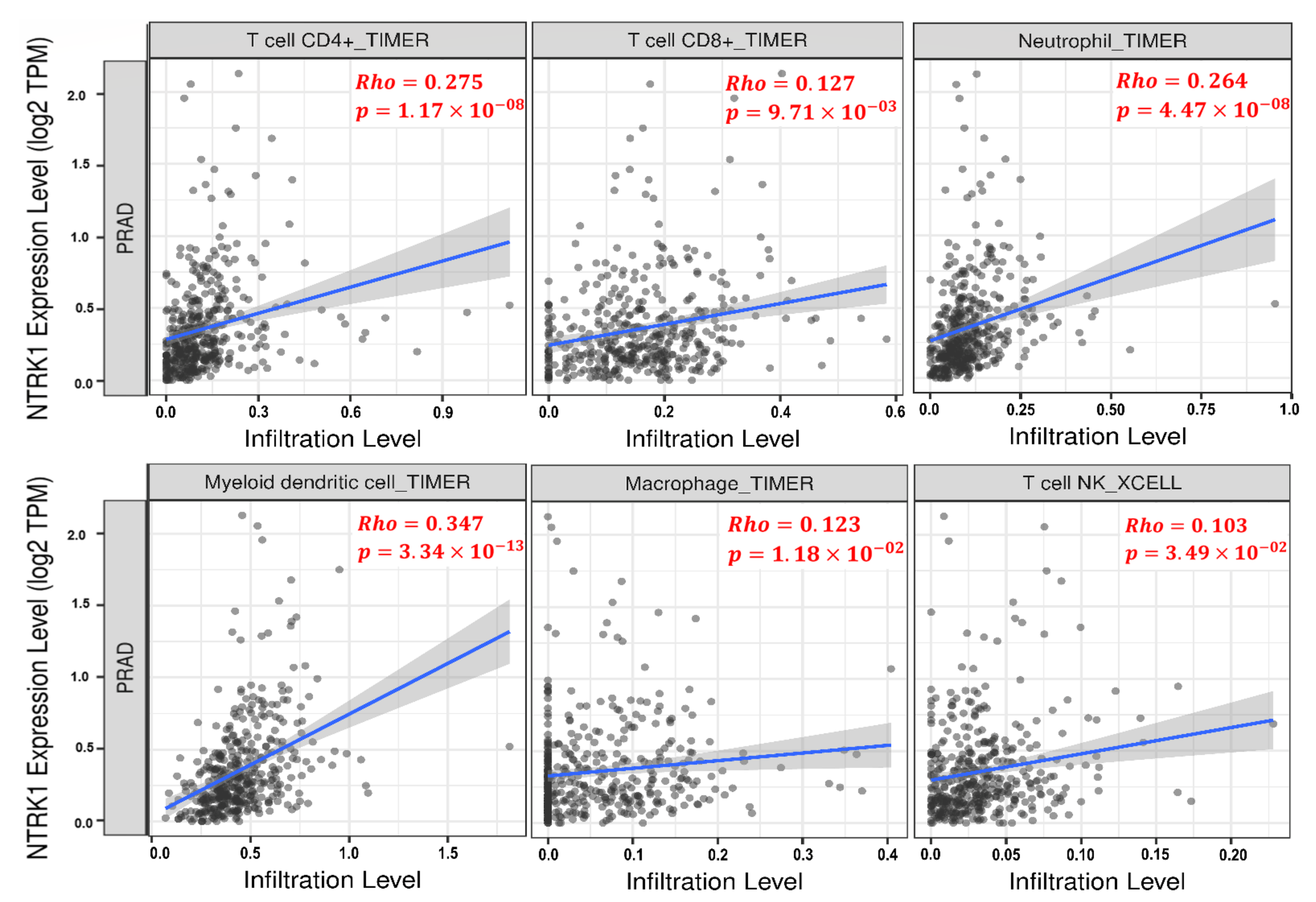
3.6. Tumor Infiltration Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Prev. Biomark. 2010, 19, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frame, F.M.; Maitland, N.J. Epigenetic Control of Gene Expression in the Normal and Malignant Human Prostate: A Rapid Response Which Promotes Therapeutic Resistance. Int. J. Mol. Sci. 2019, 20, 2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merriel, S.W.D.; Funston, G.; Hamilton, W. Prostate Cancer in Primary Care. Adv. Ther. 2018, 35, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.Z.; Zhao, X.K. Prostate cancer: Current treatment and prevention strategies. Iran. Red Crescent Med. J. 2013, 15, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.-Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef] [Green Version]
- Kohaar, I.; Petrovics, G.; Srivastava, S. A Rich Array of Prostate Cancer Molecular Biomarkers: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 1813. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-Y.; Jan, Y.-J.; Kuo, L.-K.; Wang, B.-J.; Huo, C.; Jiang, S.S.; Chen, S.-C.; Kuo, Y.-Y.; Chang, C.-R.; Chuu, C.-P. Elevation of androgen receptor promotes prostate cancer metastasis by induction of epithelial-mesenchymal transition and reduction of KAT5. Cancer Sci. 2018, 109, 3564–3574. [Google Scholar] [CrossRef]
- Liu, S.; Wang, W.; Zhao, Y.; Liang, K.; Huang, Y. Identification of Potential Key Genes for Pathogenesis and Prognosis in Prostate Cancer by Integrated Analysis of Gene Expression Profiles and the Cancer Genome Atlas. Front. Oncol. 2020, 10, 809. [Google Scholar] [CrossRef]
- Cavanagh, H.; Rogers, K.M.A. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered. Cancer Clin. Pract. 2015, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Stone, L. The IMPACT of BRCA2 in prostate cancer. Nat. Rev. Urol. 2019, 16, 639. [Google Scholar] [CrossRef]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Min, F.; Gao, F.; Liu, Z. Screening and further analyzing differentially expressed genes in acute idiopathic pulmonary fibrosis with DNA microarray. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2784–2790. [Google Scholar]
- Tan, S.H.; Petrovics, G.; Srivastava, S. Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. Int. J. Mol. Sci. 2018, 19, 1255. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Chen, S.; Ma, A.; Lu, J.; Wang, T. Identification of the difference in the pathogenesis in heart failure arising from different etiologies using a microarray dataset. Clinics 2017, 72, 600–608. [Google Scholar] [CrossRef]
- Deng, S.P.; Zhu, L.; Huang, D.S. Predicting Hub Genes Associated with Cervical Cancer through Gene Co-Expression Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 27–35. [Google Scholar] [CrossRef]
- Katoh, M.; Igarashi, M.; Fukuda, H.; Nakagama, H.; Katoh, M. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 2013, 328, 198–206. [Google Scholar] [CrossRef]
- Hong, S.K. Kallikreins as Biomarkers for Prostate Cancer. BioMed Res. Int. 2014, 2014, 526341. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.M.; Mohsen, M.T.; Malik, M.Z.; Bagabir, S.A.; Alkhanani, M.F.; Haque, S.; Serajuddin, M.; Bharadwaj, M. Identification of Potential Key Genes in Prostate Cancer with Gene Expression, Pivotal Pathways and Regulatory Networks Analysis Using Integrated Bioinformatics Methods. Genes 2022, 13, 655. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhu, Z.; Zhao, Y.; Zhang, Q.; Wu, X.; Miao, B.; Cao, J.; Fei, S. FN1, SPARC, and SERPINE1 are highly expressed and significantly related to a poor prognosis of gastric adenocarcinoma revealed by microarray and bioinformatics. Sci. Rep. 2019, 9, 7827. [Google Scholar] [CrossRef] [Green Version]
- Welsh, E.A.; Stewart, P.A.; Kuenzi, B.M.; Eschrich, J.A. Escape Excel: A tool for preventing gene symbol and accession conversion errors. PLoS ONE 2017, 12, e0185207. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.-H.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.-D. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS ONE 2017, 12, e0184129. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyot, P.; Ades, A.E.; Ouwens, M.J.N.M.; Welton, N.J. Enhanced secondary analysis of survival data: Reconstructing the data from published Kaplan-Meier survival curves. BMC Med. Res. Methodol. 2012, 12, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Oldham, M.; Horvath, S.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Geschwind, D. Identification and Removal of Outlier Samples (Illumina) Supplement for: “Functional Organization of the Transcriptome in Human Brain”. Dim (Dat1) 2021, 1, 197. [Google Scholar]
- Bong, A.H.L.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Maziveyi, M.; Alahari, S.K. Cell matrix adhesions in cancer: The proteins that form the glue. Oncotarget 2017, 8, 48471–48487. [Google Scholar] [CrossRef] [Green Version]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef]
- Abou El-Kasem, F.; Abulkheir, I.; Sidhom, N.; Ismail, A.; Habashy, H.; Elsayed, H. 275. Role of immunohistochemical expression of AMACR as a prognostic and predictive biologic marker in advanced prostatic carcinoma. Eur. J. Surg. Oncol. 2016, 42, S139. [Google Scholar] [CrossRef]
- Box, A.; Alshalalfa, M.; Hegazy, S.A.; Donnelly, B.; Bismar, T.A. High alpha-methylacyl-CoA racemase (AMACR) is associated with ERG expression and with adverse clinical outcome in patients with localized prostate cancer. Tumor Biol. 2016, 37, 12287–12299. [Google Scholar] [CrossRef]
- Gerhardt, J.; Montani, M.; Wild, P.; Beer, M.; Huber, F.; Hermanns, T.; Müntener, M.; Kristiansen, G. FOXA1 Promotes Tumor Progression in Prostate Cancer and Represents a Novel Hallmark of Castration-Resistant Prostate Cancer. Am. J. Pathol. 2012, 180, 848–861. [Google Scholar] [CrossRef]
- Teng, M.; Zhou, S.; Cai, C.; Lupien, M.; He, H.H. Pioneer of prostate cancer: Past, present and the future of FOXA1. Protein Cell 2021, 12, 29–38. [Google Scholar] [CrossRef]
- Labbé, D.P.; Brown, M. Transcriptional Regulation in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Chao, J.; Chen, L.-M.; Chai, K.X. Chapter 608—Human Kallikrein-related Peptidase 2. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 2762–2765. [Google Scholar]
- Williams, S.A.; Xu, Y.; De Marzo, A.M.; Isaacs, J.T.; Denmeade, S.R. Prostate-specific antigen (PSA) is activated by KLK2 in prostate cancer ex vivo models and in prostate-targeted PSA/KLK2 double transgenic mice. Prostate 2010, 70, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K. The role of Src in prostate cancer. Ann. Oncol. 2007, 18, 1765–1773. [Google Scholar] [CrossRef]
- Seiz, L.; Kotzsch, M.; Grebenchtchikov, N.I.; Geurts-Moespot, A.J.; Fuessel, S.; Goettig, P.; Gkazepis, A.; Wirth, M.P.; Schmitt, M.; Lossnitzer, A.; et al. Polyclonal antibodies against kallikrein-related peptidase 4 (KLK4): Immunohistochemical assessment of KLK4 expression in healthy tissues and prostate cancer. Biol. Chem. 2010, 391, 391–401. [Google Scholar] [CrossRef]
- Zhou, H.-J.; Yan, J.; Luo, W.; Ayala, G.; Lin, S.-H.; Erdem, H.; Ittmann, M.; Tsai, S.Y.; Tsai, M.-J. SRC-3 Is Required for Prostate Cancer Cell Proliferation and Survival. Cancer Res. 2005, 65, 7976–7983. [Google Scholar] [CrossRef] [Green Version]
- Groskopf, J.; Aubin, S.M.; Deras, I.L.; Blase, A.; Bodrug, S.; Clark, C.; Brentano, S.; Mathis, J.; Pham, J.; Meyer, T.; et al. APTIMA PCA3 Molecular Urine Test: Development of a Method to Aid in the Diagnosis of Prostate Cancer. Clin. Chem. 2006, 52, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Hessels, D.; van Gils, M.P.M.Q.; van Hooij, O.; Jannink, S.A.; Witjes, J.A.; Verhaegh, G.W.; Schalken, J.A. Predictive value of PCA3 in urinary sediments in determining clinico-pathological characteristics of prostate cancer. Prostate 2010, 70, 10–16. [Google Scholar] [CrossRef]
- Roobol, M.J.; Schröder, F.H.; van Leeuwen, P.; Wolters, T.; van den Bergh, R.C.N.; van Leenders, G.J.L.H.; Hessels, D. Performance of the Prostate Cancer Antigen 3 (PCA3) Gene and Prostate-Specific Antigen in Prescreened Men: Exploring the Value of PCA3 for a First-line Diagnostic Test. Eur. Urol. 2010, 58, 475–481. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Urinary biomarkers of prostate cancer. Int. J. Urol. 2018, 25, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Zhu, S.; Chen, J.; Niu, Y.; Zhou, L. A-Methylacyl-CoA Racemase (AMACR) and Prostate-Cancer Risk: A Meta-Analysis of 4,385 Participants. PLoS ONE 2013, 8, e74386. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, Y.; Zhao, J.; Zhou, W.; Wang, W.; Han, B.; Wang, X. FOXA1 promotes prostate cancer angiogenesis by inducing multiple pro-angiogenic factors expression. J. Cancer Res. Clin. Oncol. 2021, 147, 3225–3243. [Google Scholar] [CrossRef]
- Tailor, P.D.; Kodeboyina, S.K.; Bai, S.; Patel, N.; Sharma, S.; Ratnani, A.; Copland, J.A.; She, J.-X.; Sharma, A. Diagnostic and prognostic biomarker potential of kallikrein family genes in different cancer types. Oncotarget 2018, 9, 17876–17888. [Google Scholar] [CrossRef] [Green Version]
- Gunelli, R.; Fragalà, E.; Fiori, M. PCA3 in Prostate Cancer. Methods Mol. Biol. (Clifton N.J.) 2021, 2292, 105–113. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhao, Y.; Tian, Y. Comparison of diagnostic efficacy by two urine PCA3 scores in prostate cancer patients undergoing repeat biopsies. Minerva Urol. E Nefrol. = Ital. J. Urol. Nephrol. 2019, 71, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Sun, Y.; Yang, H.-L.; Zhang, B.; Wen, J.; Shi, B.-K. DLX1, a binding protein of beta-catenin, promoted the growth and migration of prostate cancer cells. Exp. Cell Res. 2018, 363, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, X.; Song, P.; Wang, H.; Wang, X. HOXC6 in the prognosis of prostate cancer. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2715–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri, S.; Rahban, M.; Bostanian, F.; Esmaeilzadeh, F.; Bagherabadi, A.; Zolghadri, S.; Stanek, A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 2022, 14, 515. [Google Scholar] [CrossRef]
- Luberg, K.; Park, R.; Aleksejeva, E.; Timmusk, T. Novel transcripts reveal a complex structure of the human TRKA gene and imply the presence of multiple protein isoforms. BMC Neurosci. 2015, 16, 78. [Google Scholar] [CrossRef] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Okamura, R.; Boichard, A.; Kato, S.; Sicklick, J.K.; Bazhenova, L.; Kurzrock, R. Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis. Oncol. 2018, 2, 1–20. [Google Scholar] [CrossRef]
- Tacconelli, A.; Farina, A.R.; Cappabianca, L.; DeSantis, G.; Tessitore, A.; Vetuschi, A.; Sferra, R.; Rucci, N.; Argenti, B.; Screpanti, I.; et al. TrkA alternative splicing: A regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004, 6, 347–360. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, A.; Farina, A.R.; Cappabianca, L.; Gulino, A.; Mackay, A.R. Alternative TrkAIII splicing: A potential regulated tumor-promoting switch and therapeutic target in neuroblastoma. Future Oncol. 2005, 1, 689–698. [Google Scholar] [CrossRef]
- Funke, L.; Bracht, T.; Oeck, S.; Schork, K.; Stepath, M.; Dreesmann, S.; Eisenacher, M.; Sitek, B.; Schramm, A. NTRK1/TrkA Signaling in Neuroblastoma Cells Induces Nuclear Reorganization and Intra-Nuclear Aggregation of Lamin A/C. Cancers 2021, 13, 5293. [Google Scholar] [CrossRef]
- Eggert, A.; Ikegaki, N.; Liu, X.-g.; Brodeur, G.M. Prognostic and Biological Role of Neurotrophin- Receptor TrkA and TrkB in Neuroblastoma. Klin Padiatr 2000, 212, 200–205. [Google Scholar] [CrossRef]
- Iraci, N.; Diolaiti, D.; Papa, A.; Porro, A.; Valli, E.; Gherardi, S.; Herold, S.; Eilers, M.; Bernardoni, R.; Valle, G.D.; et al. A SP1/MIZ1/MYCN Repression Complex Recruits HDAC1 at the TRKA and p75NTR Promoters and Affects Neuroblastoma Malignancy by Inhibiting the Cell Response to NGF. Cancer Res. 2011, 71, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Chen, H.; Luo, W.; Zhang, H.; Li, G.; Zeng, F.; Deng, F. The Landscape of Immune Cells Infiltrating in Prostate Cancer. Front. Oncol. 2020, 10, 517637. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology; Saunders/Elsevier: Philadelphia, PA, USA, 2021. [Google Scholar]
- Miller, A.M.; Lundberg, K.; Özenci, V.; Banham, A.H.; Hellström, M.; Egevad, L.; Pisa, P. CD4+CD25high T Cells Are Enriched in the Tumor and Peripheral Blood of Prostate Cancer Patients. J. Immunol. 2006, 177, 7398. [Google Scholar] [CrossRef] [Green Version]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Zidlik, V.; Brychtova, S.; Uvirova, M.; Ziak, D.; Dvorackova, J. The Changes of Angiogenesis and Immune Cell Infiltration in the Intra- and Peri-Tumoral Melanoma Microenvironment. Int. J. Mol. Sci. 2015, 16, 7876–7889. [Google Scholar] [CrossRef] [Green Version]
- Pajtler, K.W.; Rebmann, V.; Lindemann, M.; Schulte, J.; Schulte, S.; Stauder, M.; Leuschner, I.; Schmid, K.; Koehl, U.; Schramm, A.; et al. Expression of NTRK1/TrkA affects immunogenicity of neuroblastoma cells. Int. J. Cancer 2013, 133, 908–919. [Google Scholar] [CrossRef]
- Frattini, M.; Ferrario, C.; Bressan, P.; Balestra, D.; De Cecco, L.; Mondellini, P.; Bongarzone, I.; Collini, P.; Gariboldi, M.; Pilotti, S.; et al. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene 2004, 23, 7436–7440. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod. Pathol. 2019, 32, 147–153. [Google Scholar] [CrossRef]
- Sreenivasan, S.; Thirumalai, K.; Krishnakumar, S. Expression profile of genes regulated by curcumin in Y79 retinoblastoma cells. Nutr. Cancer 2012, 64, 607–616. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Symbol | Probe ID | Log2FC | Adjusted p-Value | State |
|---|---|---|---|---|
| OR51E2 | 236121_at | 4.244412 | Upregulated | |
| DLX1 | 242138_at | 3.898046 | Upregulated | |
| B3GAT1 | 219521_at | 3.883768 | Upregulated | |
| LINC00992 | 239319_at | 3.556998 | Upregulated | |
| DNASE1 | 210165_at | 3.435794 | Upregulated | |
| FFAR2 | 221345_at | 3.332304 | Upregulated | |
| FOLH1B | 211303_x_at | 3.277545 | Upregulated | |
| CLDN3 | 203954_x_at | 3.057903 | Upregulated | |
| HPN | 204934_s_at | 3.054792 | Upregulated | |
| TRPM4 | 219360_s_at | 3.014302 | Upregulated | |
| CXCL13 | 205242_at | −6.36684 | Downregulated | |
| SMTNL2 | 229730_at | −4.85304 | Downregulated | |
| SMR3B | 207441_at | −4.66651 | Downregulated | |
| FBXL21P | 1555412_at | −4.07877 | Downregulated | |
| NELL2 | 203413_at | −3.99192 | Downregulated | |
| SERPINA5 | 209443_at | −3.56837 | Downregulated | |
| BMP5 | 205431_s_at | −3.52423 | Downregulated | |
| RBP4 | 219140_s_at | −3.51651 | Downregulated | |
| FOXF2 | 206377_at | −3.49783 | Downregulated | |
| CA3 | 204865_at | −3.33564 | Downregulated |
| Genes Symbol | Probe ID | Log2FC | Adjusted p-Value | State |
|---|---|---|---|---|
| AMACR | 217111_at | 2.272572347 | Upregulated | |
| FOXA1 | 204667_at | 1.988720507 | Upregulated | |
| KLK2 | 210339_s_at | 1.577097564 | Upregulated | |
| KLK4 | 224062_x_at | 1.84883585 | Upregulated | |
| PCA3 | 232575_at | 2.979943566 | Upregulated | |
| DLX1 | 242138_at | 3.89804597 | Upregulated | |
| HOXC6 | 206858_s_at | 2.908075942 | Upregulated |
| KEGG ID | Description | Category | Gene Count | Rich Factor | BH-p-Value |
|---|---|---|---|---|---|
| has04510 | Focal adhesion | KEGG pathway | 29 | 14.42% | |
| has05414 | Dilated cardiomyopathy | KEGG pathway | 18 | 18.75% | |
| has04974 | Protein digestion and absorption | KEGG pathway | 18 | 17.47% | |
| has05410 | Hypertrophic cardiomyopathy | KEGG pathway | 16 | 17.77% | |
| has04020 | Calcium signaling pathway | KEGG pathway | 27 | 11.25% | |
| has05412 | Arrhythmogenic right ventricular cardiomyopathy | KEGG pathway | 13 | 16.88% | |
| has04512 | ECM–receptor interaction | KEGG pathway | 14 | 15.90% | |
| has04151 | PI3K-Akt signaling pathway | KEGG pathway | 33 | 9.32% | |
| has05205 | Proteoglycans in cancer | KEGG pathway | 22 | 10.73% | |
| has00330 | Arginine and proline metabolism | KEGG pathway | 9 | 17.64% |
| GO Term | Description | Category | Gene Count | Rich Factor | BH-p-Value |
|---|---|---|---|---|---|
| GO:0062023 | collagen-containing extracellular matrix | CC | 75 | 18.47% | |
| GO:0030198 | extracellular matrix organization | BP | 62 | 16.84% | |
| GO:0043062 | extracellular structure organization | BP | 62 | 16.80% | |
| GO:0005201 | extracellular matrix structural constituent | MF | 40 | 24.53% | |
| GO:0042383 | sarcolemma | CC | 30 | 22.05% | |
| GO:0005539 | glycosaminoglycan binding | MF | 39 | 17.03% | |
| GO:0060485 | mesenchyme development | BP | 43 | 15.41% | |
| GO:0048762 | mesenchymal cell differentiation | BP | 37 | 16.81% | |
| GO:0048565 | digestive tract development | BP | 28 | 20.89% | . |
| GO:0042692 | muscle cell differentiation | BP | 50 | 12.98% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagherabadi, A.; Hooshmand, A.; Shekari, N.; Singh, P.; Zolghadri, S.; Stanek, A.; Dohare, R. Correlation of NTRK1 Downregulation with Low Levels of Tumor-Infiltrating Immune Cells and Poor Prognosis of Prostate Cancer Revealed by Gene Network Analysis. Genes 2022, 13, 840. https://doi.org/10.3390/genes13050840
Bagherabadi A, Hooshmand A, Shekari N, Singh P, Zolghadri S, Stanek A, Dohare R. Correlation of NTRK1 Downregulation with Low Levels of Tumor-Infiltrating Immune Cells and Poor Prognosis of Prostate Cancer Revealed by Gene Network Analysis. Genes. 2022; 13(5):840. https://doi.org/10.3390/genes13050840
Chicago/Turabian StyleBagherabadi, Arash, Amirreza Hooshmand, Nooshin Shekari, Prithvi Singh, Samaneh Zolghadri, Agata Stanek, and Ravins Dohare. 2022. "Correlation of NTRK1 Downregulation with Low Levels of Tumor-Infiltrating Immune Cells and Poor Prognosis of Prostate Cancer Revealed by Gene Network Analysis" Genes 13, no. 5: 840. https://doi.org/10.3390/genes13050840
APA StyleBagherabadi, A., Hooshmand, A., Shekari, N., Singh, P., Zolghadri, S., Stanek, A., & Dohare, R. (2022). Correlation of NTRK1 Downregulation with Low Levels of Tumor-Infiltrating Immune Cells and Poor Prognosis of Prostate Cancer Revealed by Gene Network Analysis. Genes, 13(5), 840. https://doi.org/10.3390/genes13050840