Genome-Wide Identification of the SAMS Gene Family in Upland Cotton (Gossypium hirsutum L.) and Expression Analysis in Drought Stress Treatments

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Identification of SAMS Proteins
2.2. Analysis of Phylogenetic, Gene Structure and Conserved Motif Characteristics
2.3. Chromosomal Location, Collinearity and Promoter Analysis
2.4. Digital Expression Analysis of SAMs Genes in Cotton
2.5. Drought Treatment and PCR (qRT-PCR) Analysis of Cotton Drought Resistant Materials
2.6. Protein Interaction Network Analysis of GhSAMS
3. Results
3.1. Identification of SAMS Genes in Upland Cotton
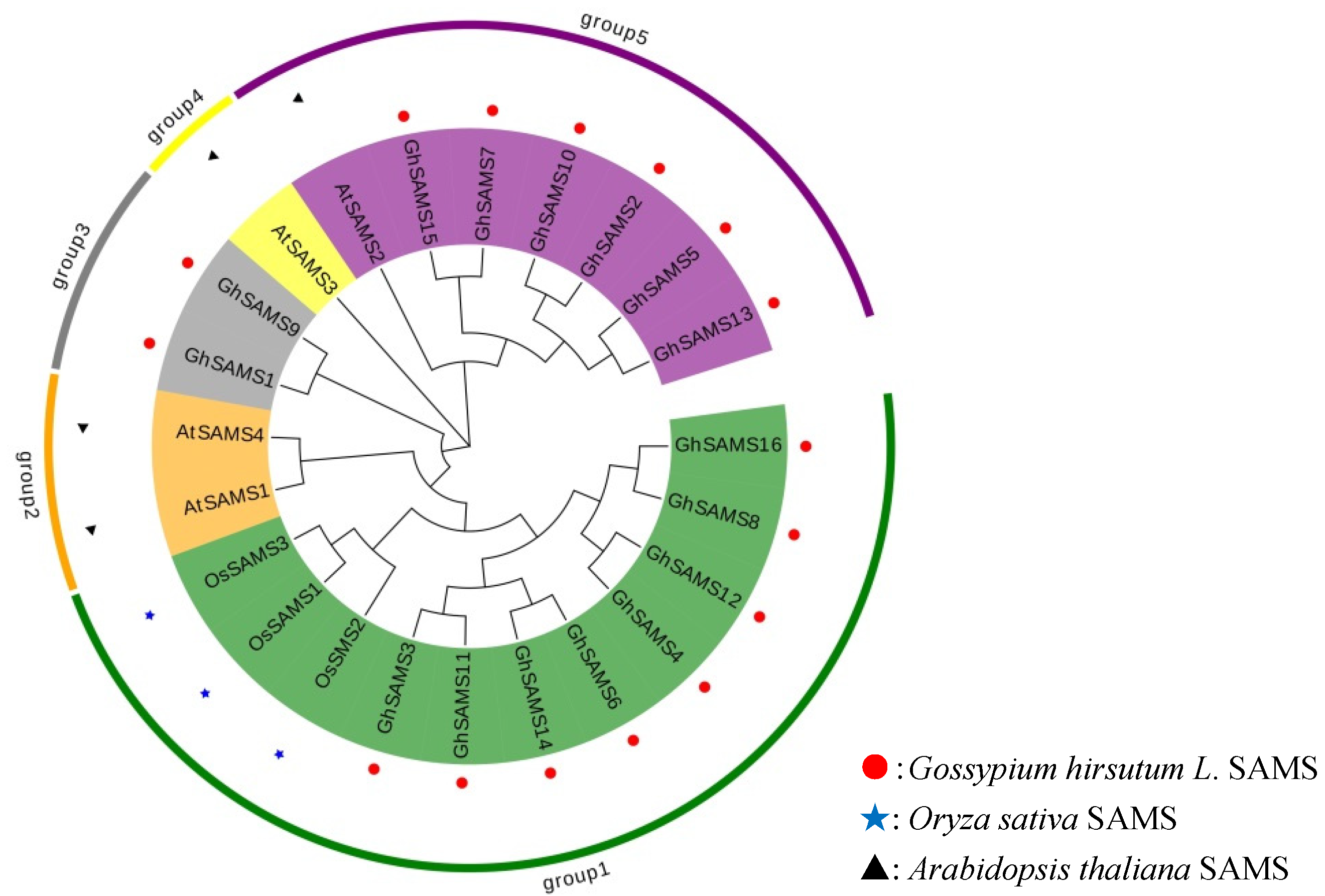
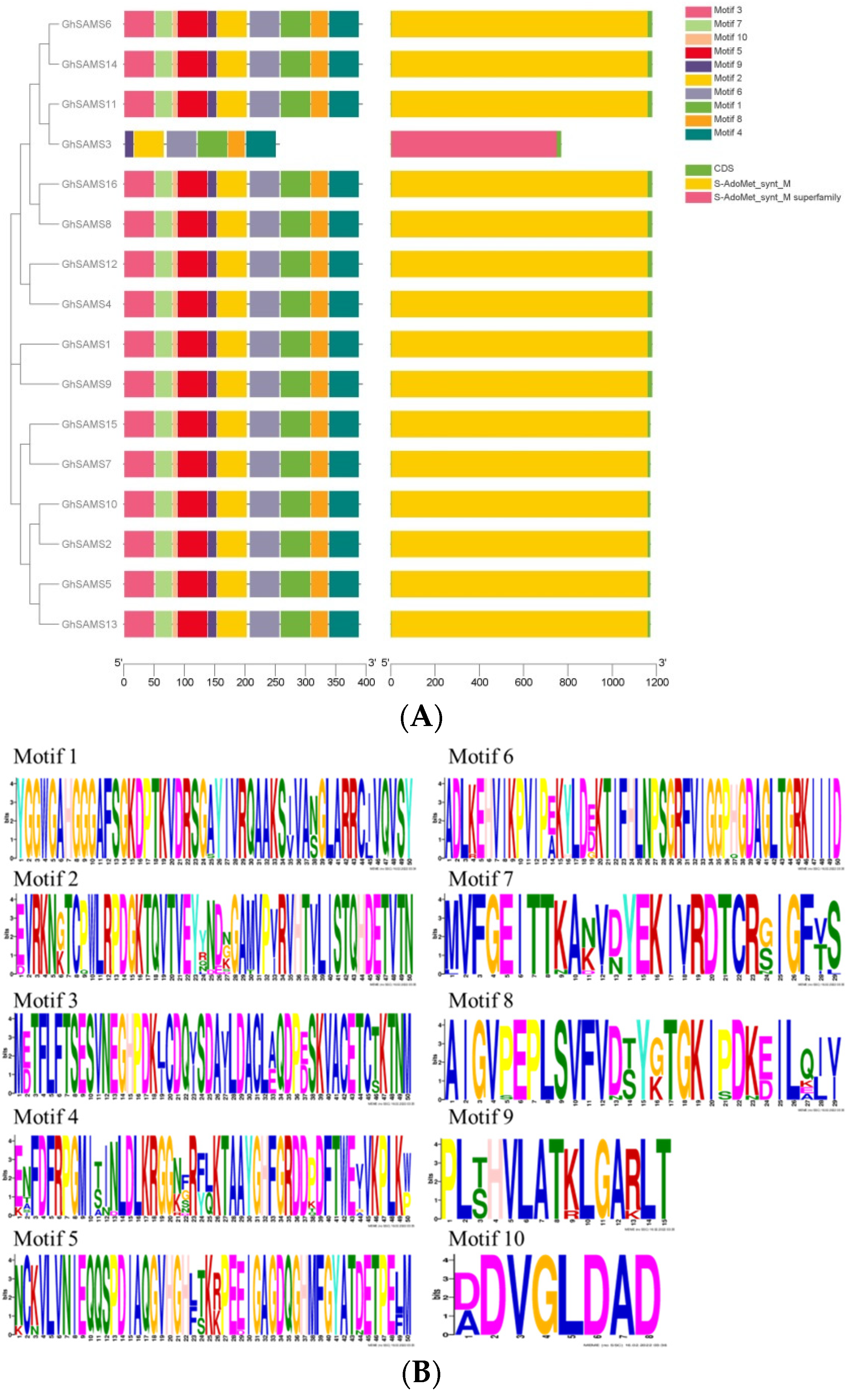
3.2. Phylogenetic, Gene Structure and Motif Identification of Upland Cotton SAMS Genes
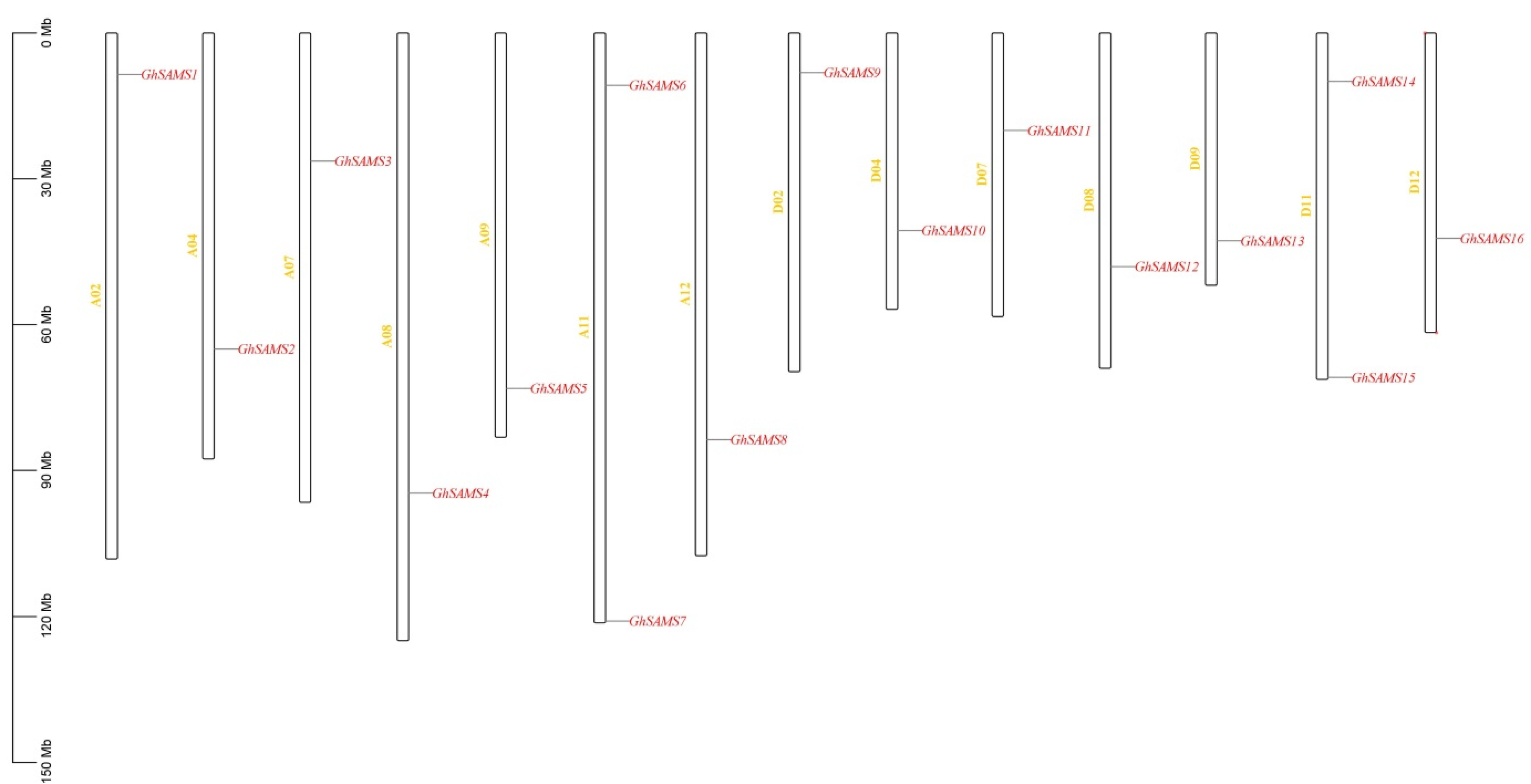
3.3. Chromosomal Location, Collinearity and Promoter Analysis of the SAMS Genes of Upland Cotton
3.4. Expression Analysis of SAMS Genes in Upland Cotton
3.5. Expression Analysis of GhSAMS after Drought Stress
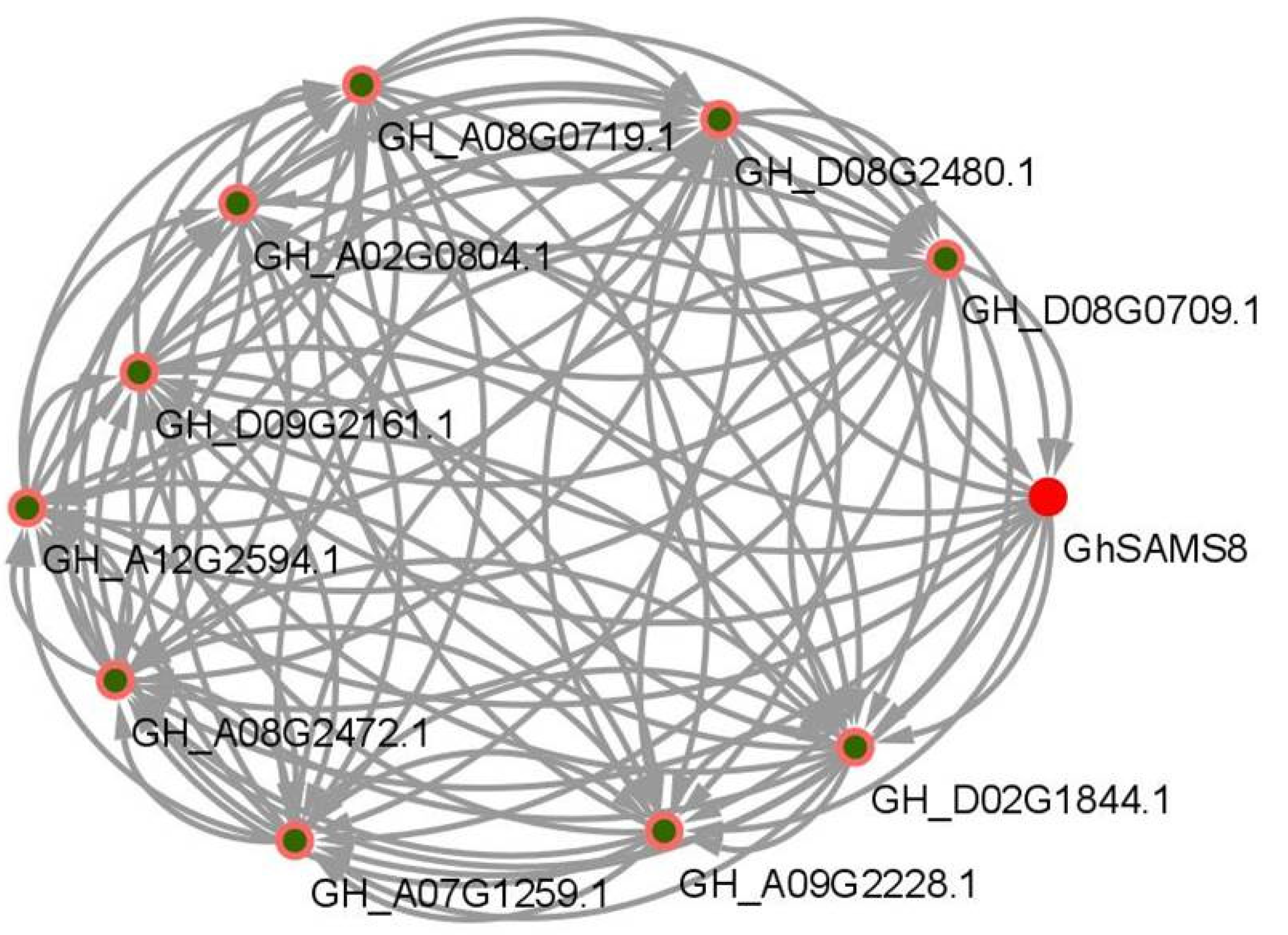
3.6. GhSAMS Protein Interaction Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, C.Y.; Yang, Z.Y.; Hu, Y.G. Drought resistance of wheat alien chromosome addition lines evaluated by membership function value based on multiple traits and drought resistance index of grain yield. Field Crops Res. 2015, 179, 103–112. [Google Scholar] [CrossRef]
- Nagy, Z.; Németh, E.; Guóth, A.; Bona, L.; Wodala, B.; Pécsváradi, A. Metabolic indicators of drought stress tolerance in wheat: Glutamine synthetase isoenzymes and Rubisco. Plant Physiol. Biochem. 2013, 67, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Fang, H.; Guo, F.; Li, F.; Chen, A.; Huang, S. Flavonoids as inducers of white adipose tissue browning and thermogenesis: Signalling pathways and molecular triggers. Nutr. Metab. 2019, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.A.; Markham, G.D. Methionine adenosyltransferase (S-adenosylmethionine synthetase). Adv. Enzym. Relat. Areas Mol. Biol. 2011, 78, 449–521. [Google Scholar]
- Gallardo, K.; Job, C.; Groot, S.P.; Puype, M.; Demol, H.; Vandekerckhove, J.; Job, D. Importance of methionine biosynthesis for Arabidopsis seed germination and seedling growth. Physiol. Plant. 2002, 116, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aguayo, I.; Rodriguez-Galan, J.M.; Garcia, R.; Torreblanca, J.; Pardo, J.M. Salt stress enhances xylem development and expression of S-adenosyl-L-methionine synthase in lignifying tissues of tomato plants. Planta 2004, 220, 278–285. [Google Scholar] [CrossRef]
- Jone, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Loraine, A.E.; Mccormick, S.; Estrada, A.; Patel, K.; Qin, P. RNA-Seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol. 2013, 162, 1092–1109. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zou, T.; Mccormick, S. S-adenosylmethionine synthetase 3 is important for pollen tube growth. Plant Physiol. 2016, 172, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Li, W.X.; Han, Y.Y.; Tao, F.; Chong, K. Knockdown of SAMS genes encoding S-adenosyl-L-methionine synthetases causes methylation alterations of DNAs and histones and leads to late flowering in rice. J. Plant Physiol. 2011, 168, 1837–1843. [Google Scholar] [CrossRef]
- Espartero, J.; Pintor-Toro, J.A.; Pardo, J.M. Differential accumulation of S-adenosylmethionine synthetase transcripts in response to salt stress. Plant Mol. Biol. 1994, 25, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Du, C.X.; Fan, H.F.; Guo, S.R.; Tezukab, T.; Li, J. Proteomic analysis of cucumber seedling roots subjected to salt stress. Phytochemistry 2010, 71, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; He, L.Z.; Guo, S.R.; Li, J.; Yang, Y.J.; Yan, B.; Sun, J.; Li, J. Proteomics reveal cucumber Spd-responses under normal condition and salt stress. Plant Physiol. Biochem. 2013, 67, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.W.; Komatsu, S. Characterization of proteins in soybean roots under flooding and drought stresses. J. Proteom. 2015, 114, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Oh, M.W.; Komatsu, S. Characterization of S-adenosylmethionine syntiietases in soybean under flooding and drought stresses. Biologia Plantarum. 2016, 60, 69–278. [Google Scholar] [CrossRef]
- Yang, Z.E.; Gong, Q.; Qin, W.Q.; Yang, Z.R.; Cheng, Y.; Lu, L.L.; Ge, X.Y.; Zhang, C.J.; Wu, Z.X.; Li, F.G. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Lingwan, M.; Yadukrishnan, P.; Masakapalli, S.K.; Datta, S. BBX31 promotes hypocotyl growth, primary root elongation and UV-B tolerance in Arabidopsis. Plant Signal. Behav. 2019, 14, e1588672. [Google Scholar] [CrossRef]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Zu, Q.L.; Qu, Y.Y.; Ni, Z.Z.; Zheng, K.; Chen, Q.; Chen, Q.J. The Chalcone Isomerase Family in Cotton: Whole-Genome Bioinformatic and Expression Analyses of the Gossypium barbadense L. Response to FusariumWilt Infection. Genes. 2019, 10, 1006. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Berri, S.; Abbruscato, P.; Faivre-Rampant, O.; Brasileiro, A.C.; Fumasoni, I.; Satoh, K.; Kikuchi, S.; Mizzi, L.; Morandini, P.; Pè, M.E.; et al. Characterization of WRKY co-regulatory networks in rice and Arabidopsis. BMC Plant Biol. 2009, 9, 120. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2014, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Baker, M.E.; Elkan, C.P. An artificial intelligence approach to motif discovery in protein sequences: Application to steroid dehydrogenases. J. Steroid Biochem. Mol. Biol. 1997, 62, 29–44. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Zhang, H.; Ma, L.; Wang, Z.; Wang, K. Genome-Wide Identification and Expression Profiling Analysis of the Trihelix Gene Family under Abiotic Stresses in Medicago truncatula. Genes. 2020, 11, 1389. [Google Scholar] [CrossRef] [PubMed]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Qu, Y.; Chen, Q.; Gao, W. Comprehensive evaluation of cotton drought tolerance indexes and gray relational analysis. Agric. Res. Arid Areas. 2019, 37, 233–239. [Google Scholar]
- Wang, W.; Cheng, Y.; Chen, D.; Liu, D.; Hu, M.; Dong, J.; Zhang, X.; Song, L.; Shen, F. The Catalase Gene Family in Cotton: Genome-Wide Characterization and Bioinformatics Analysis. Cells 2019, 8, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.Y.; Wang, P.; Gao, W.J.; Long, Y.L.; Wang, Y.X.; Geng, S.W.; Su, X.N.; Jiao, Y.; Chen, Q.J.; Qu, Y.Y. Genome-wide identification of the DUF668 gene family in cotton and expression profiling analysis of GhDUF668 in Gossypium hirsutum under adverse stress. BMC Genomics. 2021, 22, 395. [Google Scholar] [CrossRef]
- Heidari, P.; Mazloomi, F.; Nussbaumer, T.; Barcaccia, G. Insights into the SAM Synthetase Gene Family and Its Roles in Tomato Seedlings under Abiotic Stresses and Hormone Treatments. Plants 2020, 9, 586. [Google Scholar] [CrossRef]
- Sauter, M.; Moffatt, B.; Saechao, M.C.; Hell, R.; Wirtz, M. Methionine salvage and S-adenosylmethionine: Essential links between sulfur, ethylene and polyamine biosynthesis. Biochem. J. 2013, 451, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belak, Z.R.; Pickering, J.A.; Gillespie, Z.E.; Audette, G.F.; Eramian, M.; Mitchell, J.A.; Bridger, J.M.; Kusalik, A.; Eskiw, C.H. Genes responsive to rapamycin and serum deprivation are clustered on chromosomes and undergo re-organization within local chromatin environments. Biochem. Cell Biol. 2020, 98, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.H.; Tang, C.R.; Fang, Y.J.; Yang, M.; Zhou, B.H.; Qi, J.Y.; Zhang, Y. Structure and expression profile of the sucrose synthase gene family in the rubber tree: Indicative of roles in stress response and sucrose utilization in the laticifers. FEBS J. 2014, 281, 291–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murat, F.; Xu, J.H.; Tannier, E.; Abrouk, M.; Guilhot, N.; Pont, C.; Messing, J.; Salse, J. Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Res. 2012, 20, 1545–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and heads) of functional diversity. Physiology 2005, 20, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Otto, S.P.; Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [Green Version]
- Adams, K.L. Evolution of duplicate gene expression in polyploid and hybrid plants. J. Hered. 2007, 98, 136–141. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.C.; Hsieh, E.J.; Chen, J.H.; Chen, H.Y.; Lin, T.P. Arabidopsis RGLG2, functioning as a RING E3 ligase, interacts with AtERF53 and negatively regulates the plant drought stress response. Plant Physiol. 2012, 158, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.G.; Zhang, W.K.; He, S.J.; Zhang, J.S.; Liu, Q.; Chen, S.Y. An EREBP/AP2-type protein in Triticum aestivum was a DRE-binding transcription factor induced by cold, dehydration and ABA stress. Theor. Appl. Genet. 2003, 106, 923–930. [Google Scholar] [CrossRef]
- Leng, P.; Zhao, J. Transcription factors as molecular switches to regulate drought adaptation in maize. Theor. Appl. Genet. 2020, 133, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, E.; Genga, A. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NIPGR ID. | Cotton Identifier | Chromosome | Start Position | End Position | Protein (aa) | pI | Molecular Weight (Da) | CDS (bp) | Predicted Subcellular Location | GRAVY |
|---|---|---|---|---|---|---|---|---|---|---|
| GhSAMS1 | GH_A02G0616.1 | AD1-A02 | 8,514,170 | 8,515,351 | 393 | 5.680 | 43,060.86 | 1182 | Cytoplasmic | −0.308 |
| GhSAMS2 | GH_A04G0904.1 | AD1-A04 | 65,039,231 | 65,040,403 | 390 | 6.510 | 42,854.92 | 1173 | Cytoplasmic | −0.326 |
| GhSAMS3 | GH_A07G1419.1 | AD1-A07 | 26,342,235 | 26,343,005 | 256 | 8.730 | 28,120.07 | 771 | Cytoplasmic | −0.326 |
| GhSAMS4 | GH_A08G1451.1 | AD1-A08 | 94,661,115 | 94,662,296 | 393 | 5.590 | 43,090.84 | 1182 | Cytoplasmic | −0.325 |
| GhSAMS5 | GH_A09G1633.1 | AD1-A09 | 73,132,352 | 73,133,524 | 390 | 6.640 | 42,609.68 | 1173 | Cytoplasmic | −0.299 |
| GhSAMS6 | GH_A11G1106.1 | AD1-A11 | 10,728,104 | 10,729,285 | 393 | 5.480 | 43,025.52 | 1182 | Cytoplasmic | −0.360 |
| GhSAMS7 | GH_A11G3695.1 | AD1-A11 | 120,911,380 | 120,912,552 | 390 | 6.320 | 42,681.58 | 1173 | Cytoplasmic | −0.332 |
| GhSAMS8 | GH_A12G1381.1 | AD1-A12 | 83,660,094 | 83,661,275 | 393 | 5.490 | 43,070.68 | 1182 | Cytoplasmic | −0.335 |
| GhSAMS9 | GH_D02G0631.1 | AD1-D01 | 8,142,070 | 8,143,251 | 393 | 5.650 | 43,043.83 | 1182 | Cytoplasmic | −0.300 |
| GhSAMS10 | GH_D04G1227.1 | AD1-D04 | 40,645,773 | 40,646,945 | 390 | 6.500 | 42,811.94 | 1173 | Cytoplasmic | −0.306 |
| GhSAMS11 | GH_D07G1411.1 | AD1-D07 | 20,066,986 | 20,068,167 | 393 | 5.490 | 43,038.68 | 1182 | Cytoplasmic | −0.328 |
| GhSAMS12 | GH_D08G1473.1 | AD1-D08 | 48,053,425 | 48,054,606 | 393 | 5.520 | 43,039.75 | 1182 | Cytoplasmic | −0.338 |
| GhSAMS13 | GH_D09G1576.1 | AD1-D09 | 42,758,795 | 42,759,967 | 390 | 6.640 | 42,694.79 | 1173 | Cytoplasmic | −0.294 |
| GhSAMS14 | GH_D11G1137.1 | AD1-D11 | 9,955,464 | 9,956,645 | 393 | 5.490 | 43,061.63 | 1182 | Cytoplasmic | −0.358 |
| GhSAMS15 | GH_D11G3723.1 | AD1-D11 | 70,868,476 | 70,869,648 | 390 | 6.320 | 42,615.56 | 1173 | Cytoplasmic | −0.320 |
| GhSAMS16 | GH_D12G1397.1 | AD1-D12 | 42,290,014 | 42,291,195 | 393 | 5.490 | 43,041.68 | 1182 | Cytoplasmic | −0.321 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, F.; Ma, J.; Wang, P.; Yang, Y. Genome-Wide Identification of the SAMS Gene Family in Upland Cotton (Gossypium hirsutum L.) and Expression Analysis in Drought Stress Treatments. Genes 2022, 13, 860. https://doi.org/10.3390/genes13050860
Sun F, Ma J, Wang P, Yang Y. Genome-Wide Identification of the SAMS Gene Family in Upland Cotton (Gossypium hirsutum L.) and Expression Analysis in Drought Stress Treatments. Genes. 2022; 13(5):860. https://doi.org/10.3390/genes13050860
Chicago/Turabian StyleSun, Fenglei, Jun Ma, Penglong Wang, and Yanlong Yang. 2022. "Genome-Wide Identification of the SAMS Gene Family in Upland Cotton (Gossypium hirsutum L.) and Expression Analysis in Drought Stress Treatments" Genes 13, no. 5: 860. https://doi.org/10.3390/genes13050860