
Identification of the circRNA-miRNA-mRNA Prognostic Regulatory Network in Lung Adenocarcinoma

Abstract
:1. Introduction
2. Methods
2.1. Source of LUAD Datasets
2.2. Differential circRNA Expression and Venn Analysis
2.3. Definition of the miRNAs-Related Prognostic Model
2.4. Evaluation of the Risk Model and Identification of Potential Biomarkers
2.5. Prediction of miRNA Binding Sites and Target mRNAs
2.6. GO and KEGG Pathway Analyses
2.7. Establishment of PPI Network and Identification of Hub-Genes
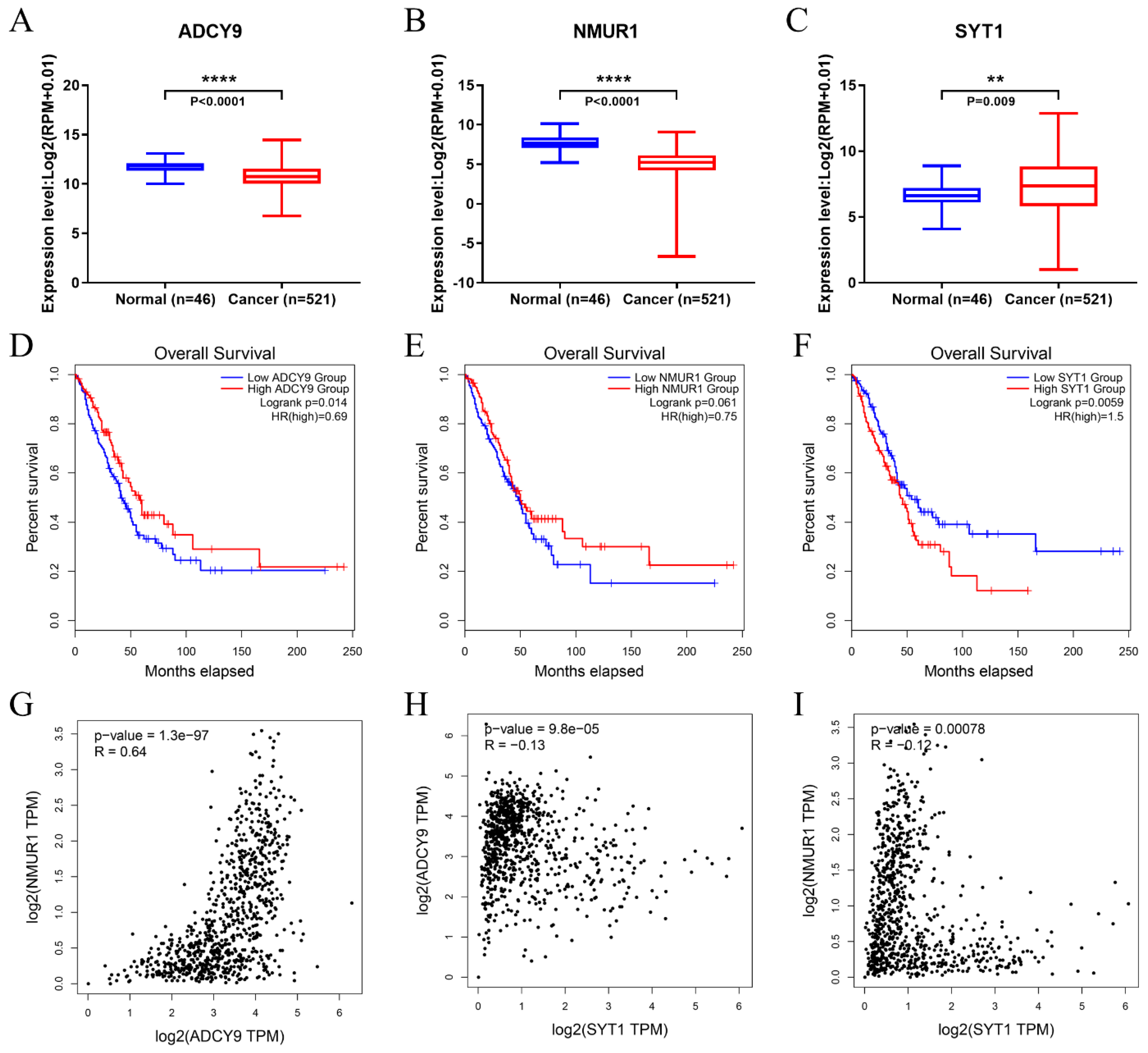
2.8. Differentially Expressed and OS Analysis of the Hub Genes
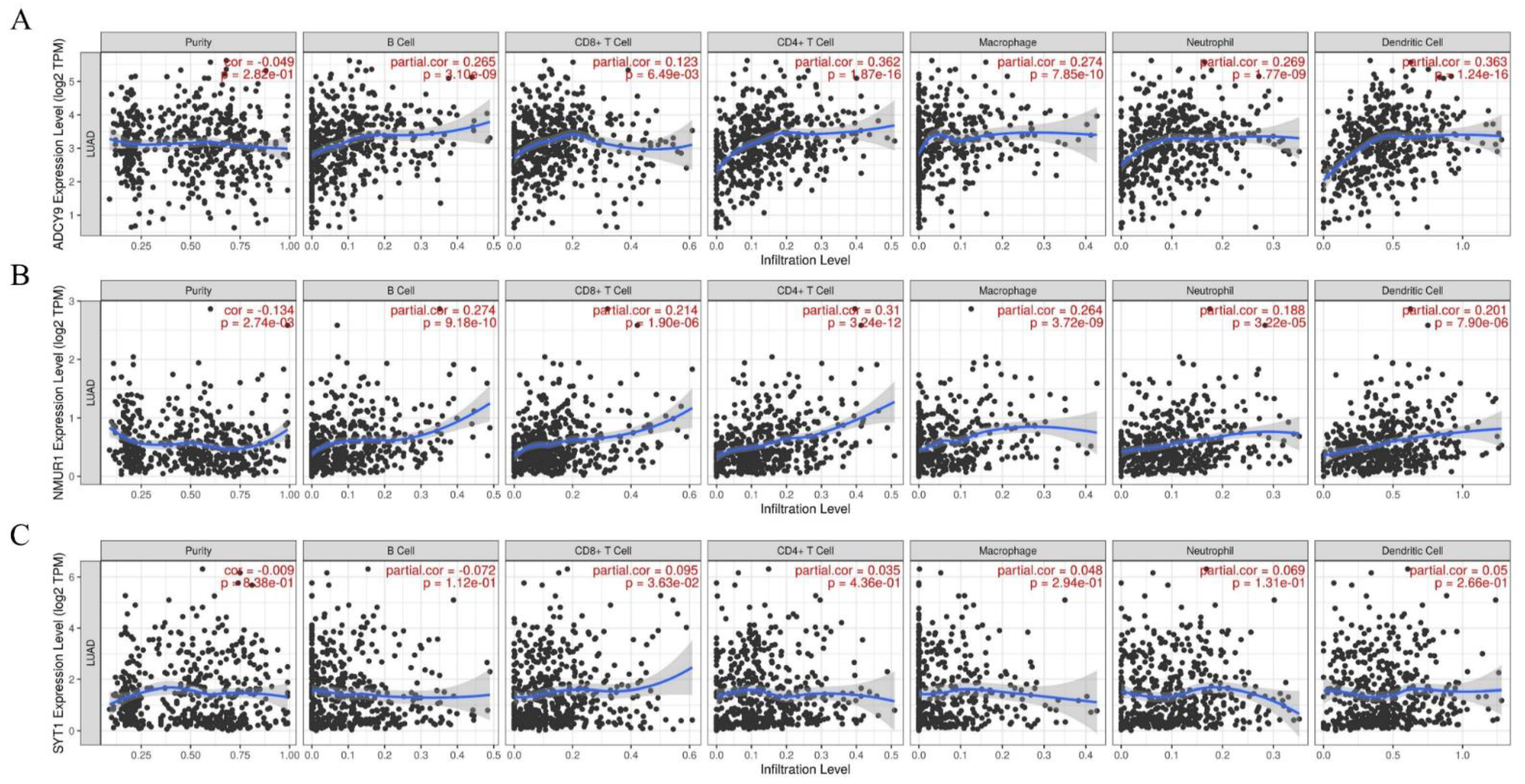
2.9. Correlation Analysis of Hub Gene and Identification of the Association between Hub Gene and Immune Infiltration in LUAD
3. Results
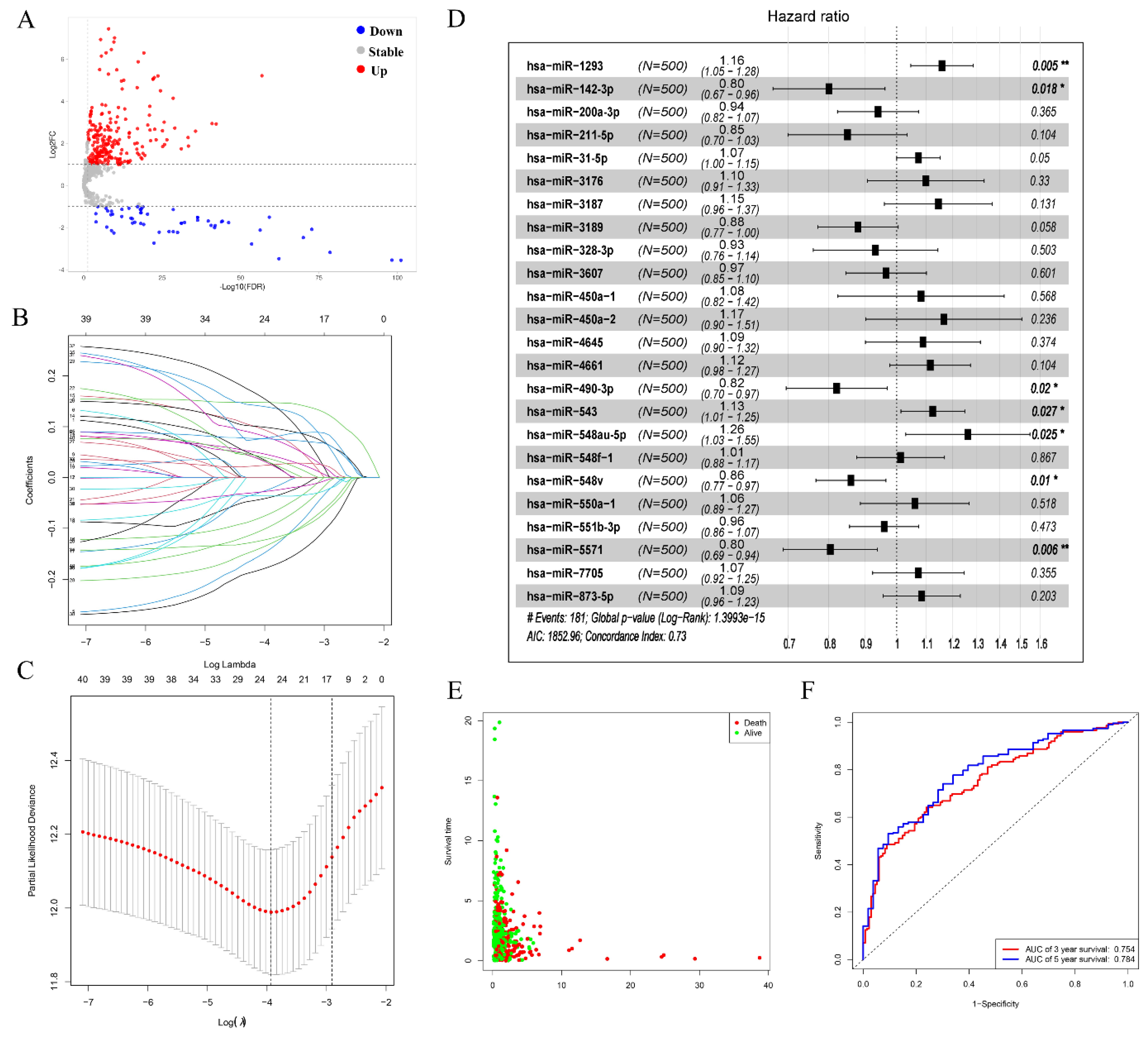
3.1. Differentially Expressed circRNAs Analysis in the NSCLC
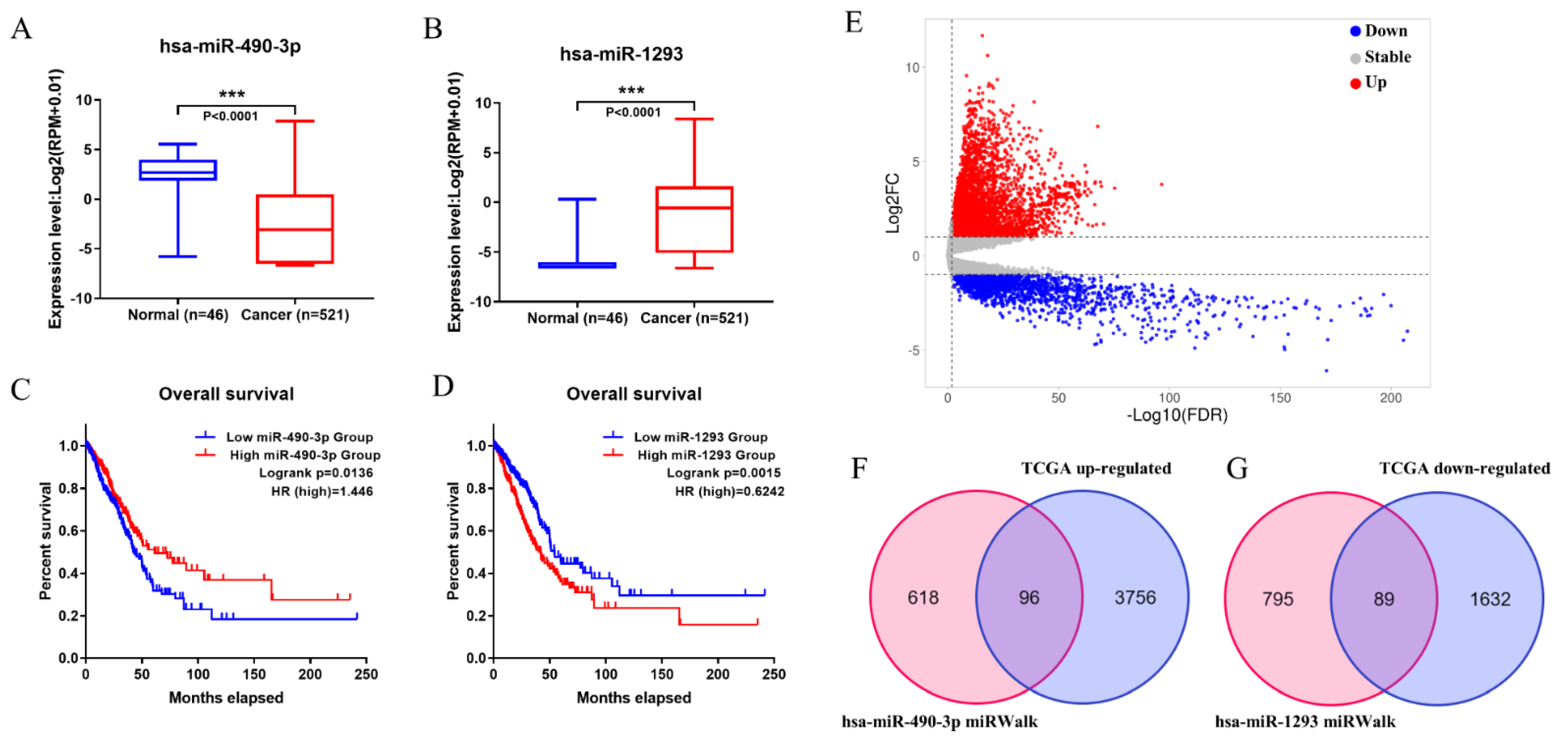
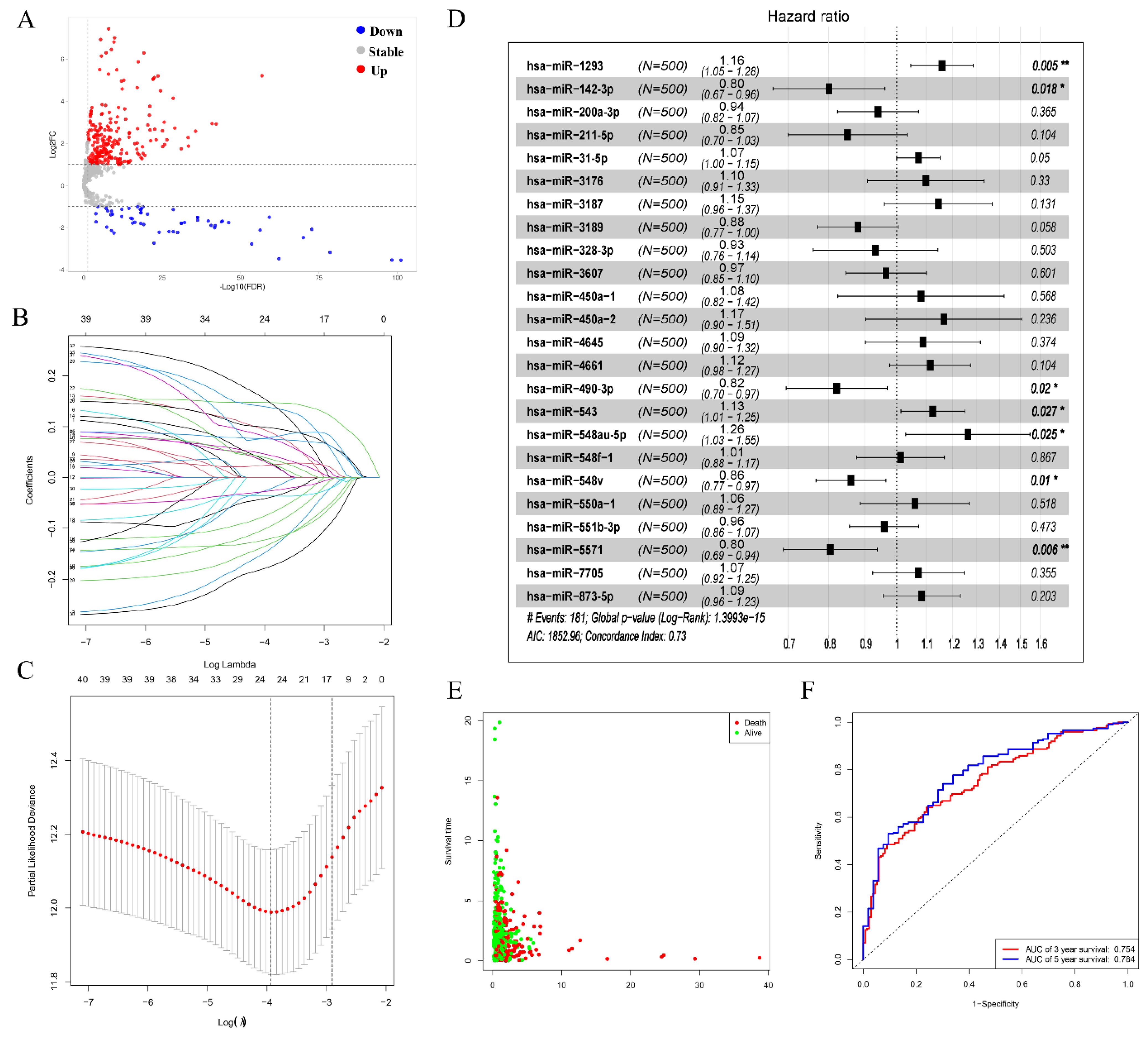
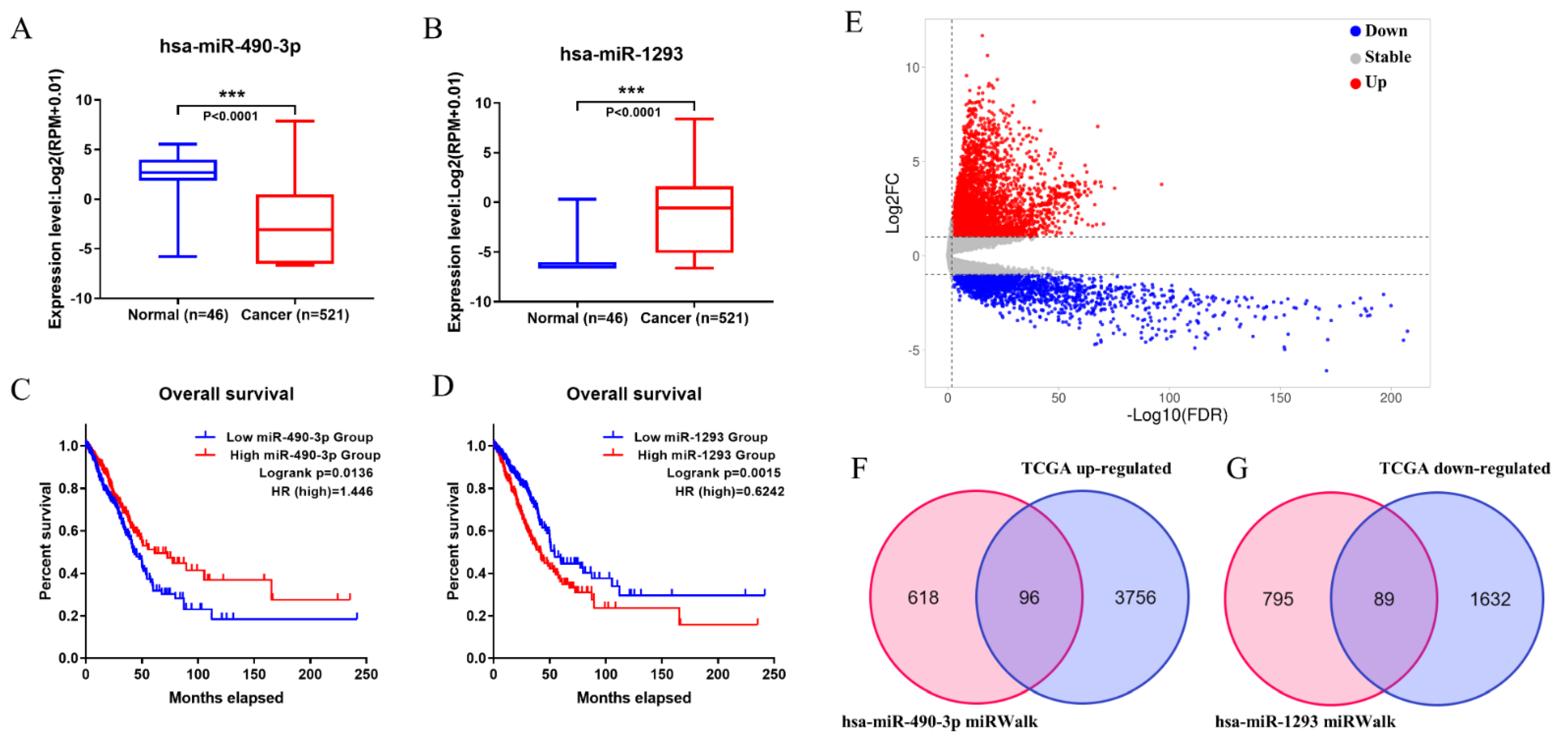
3.2. Prognostic-Related Differential miRNAs from TCGA
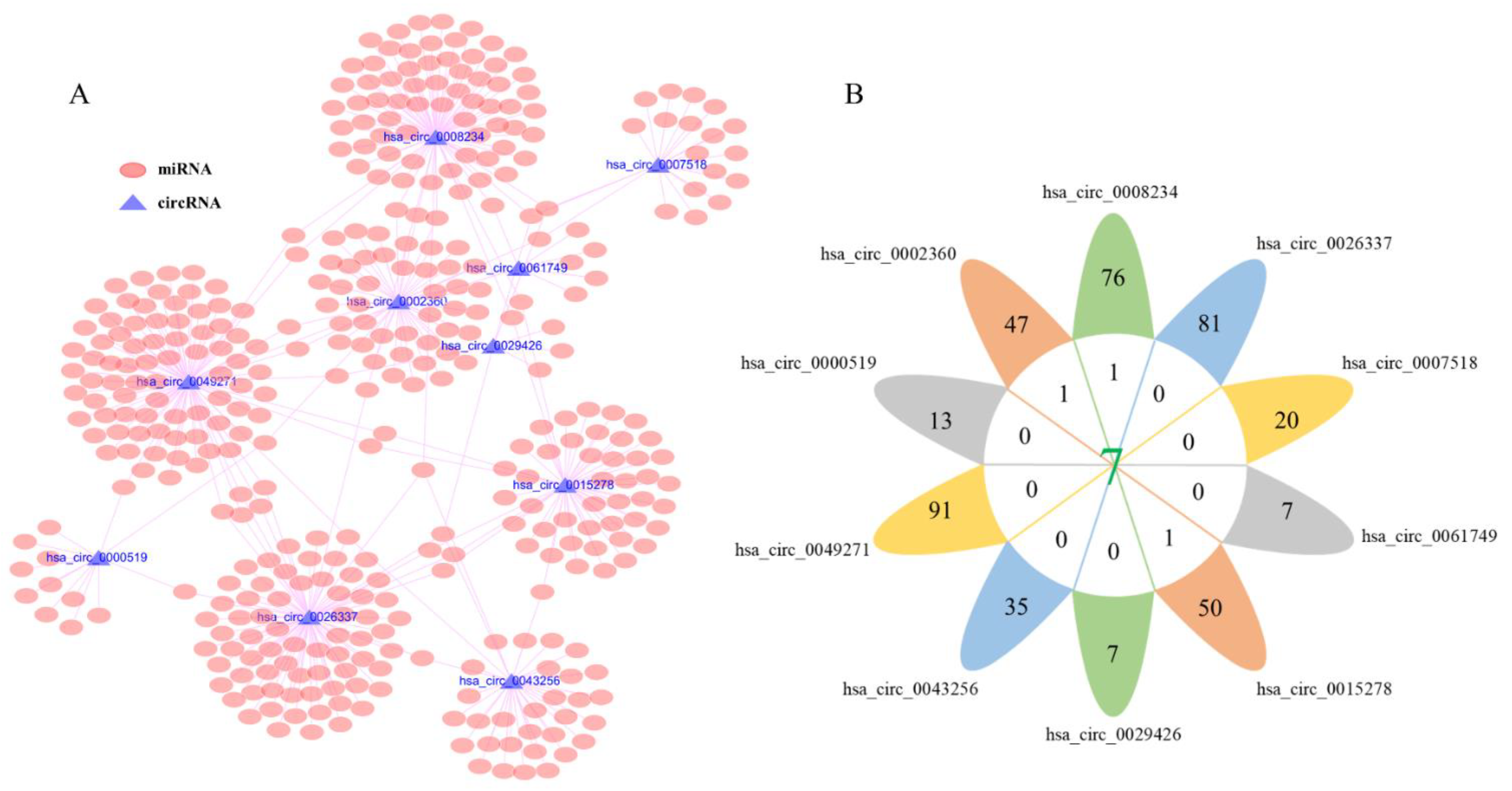
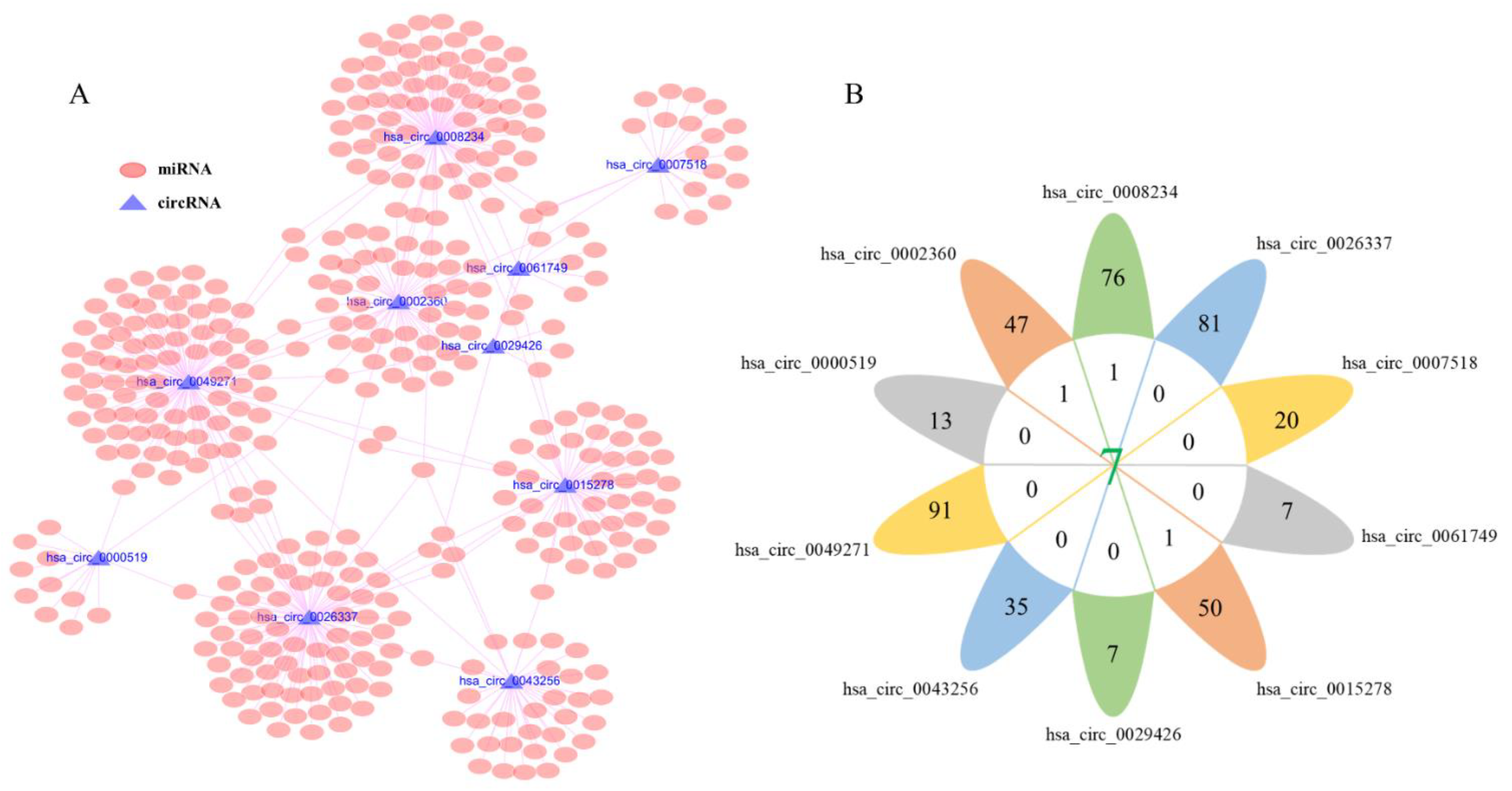
3.3. Prediction of Target miRNAs and Identification of circRNA-miRNA Interactions
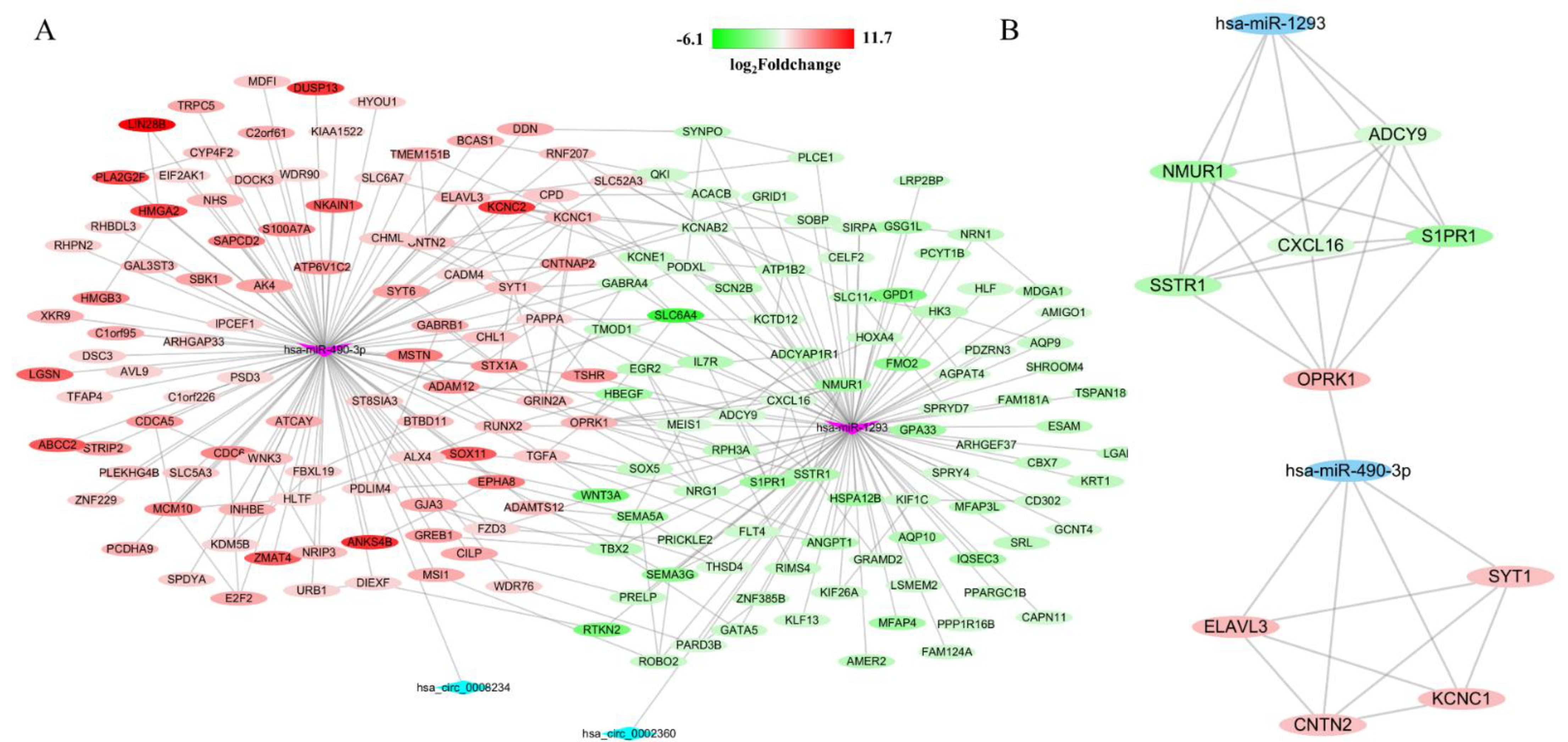
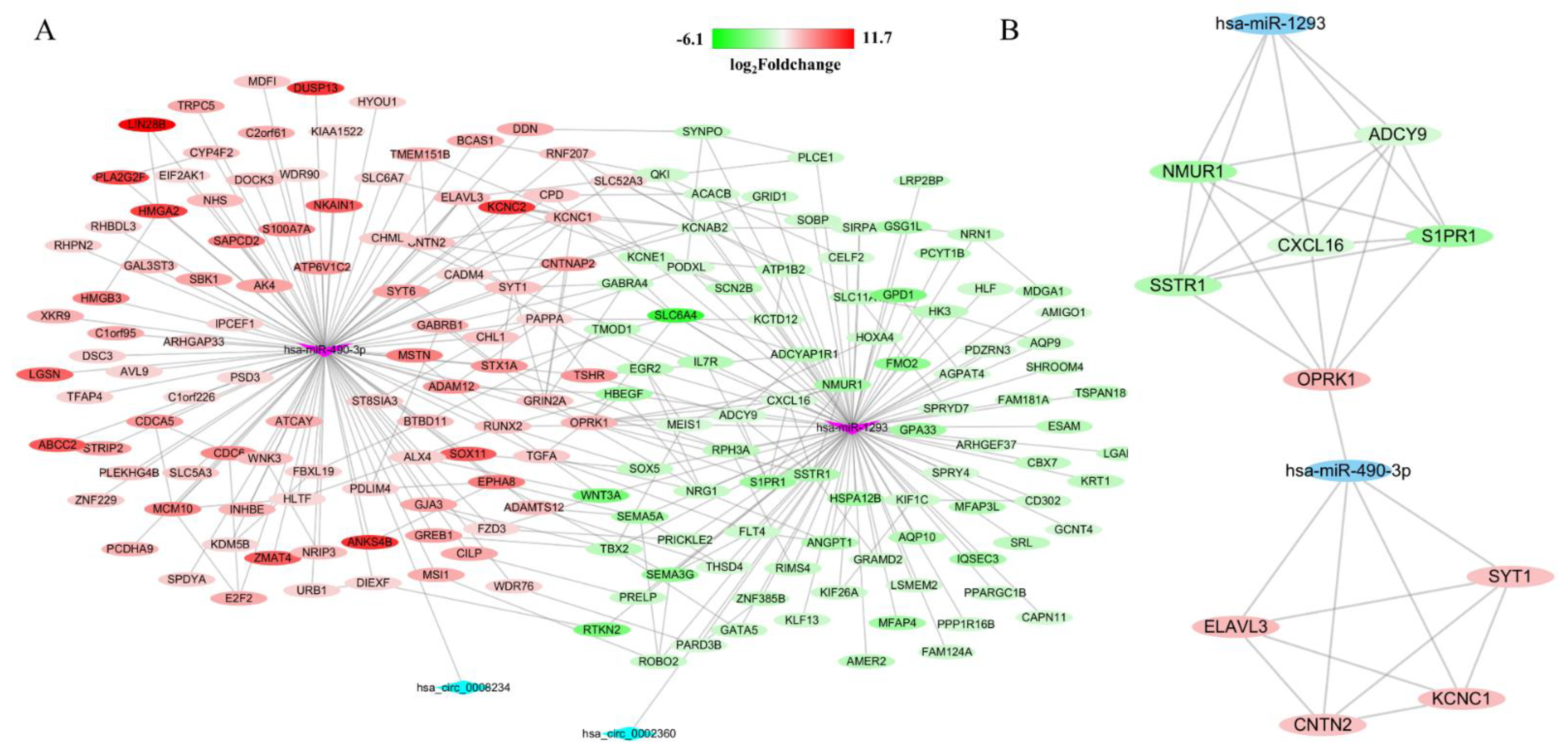
3.4. Construction of circRNA-miRNA-mRNA Network
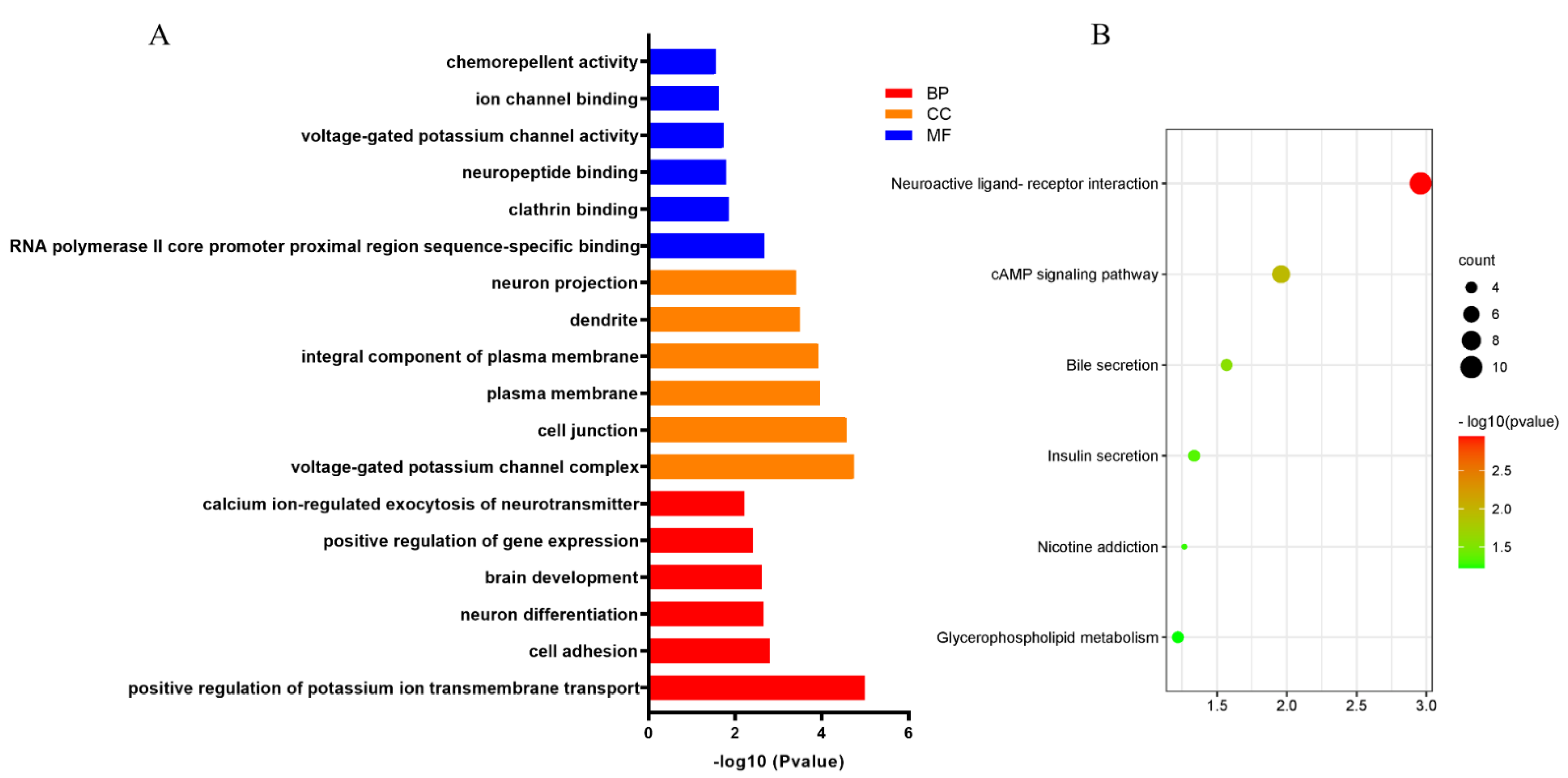
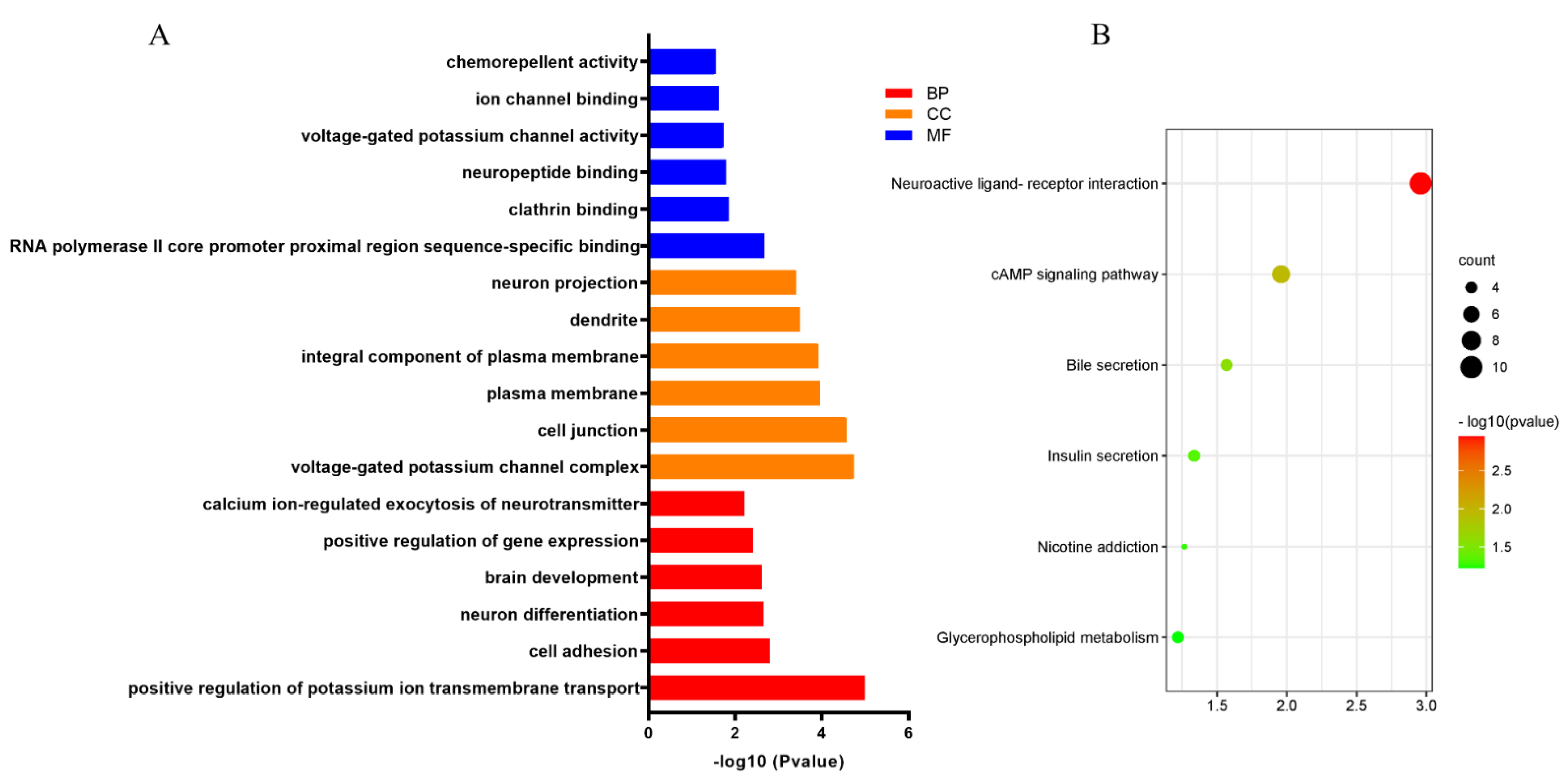
3.5. Functional Enrichment Analysis
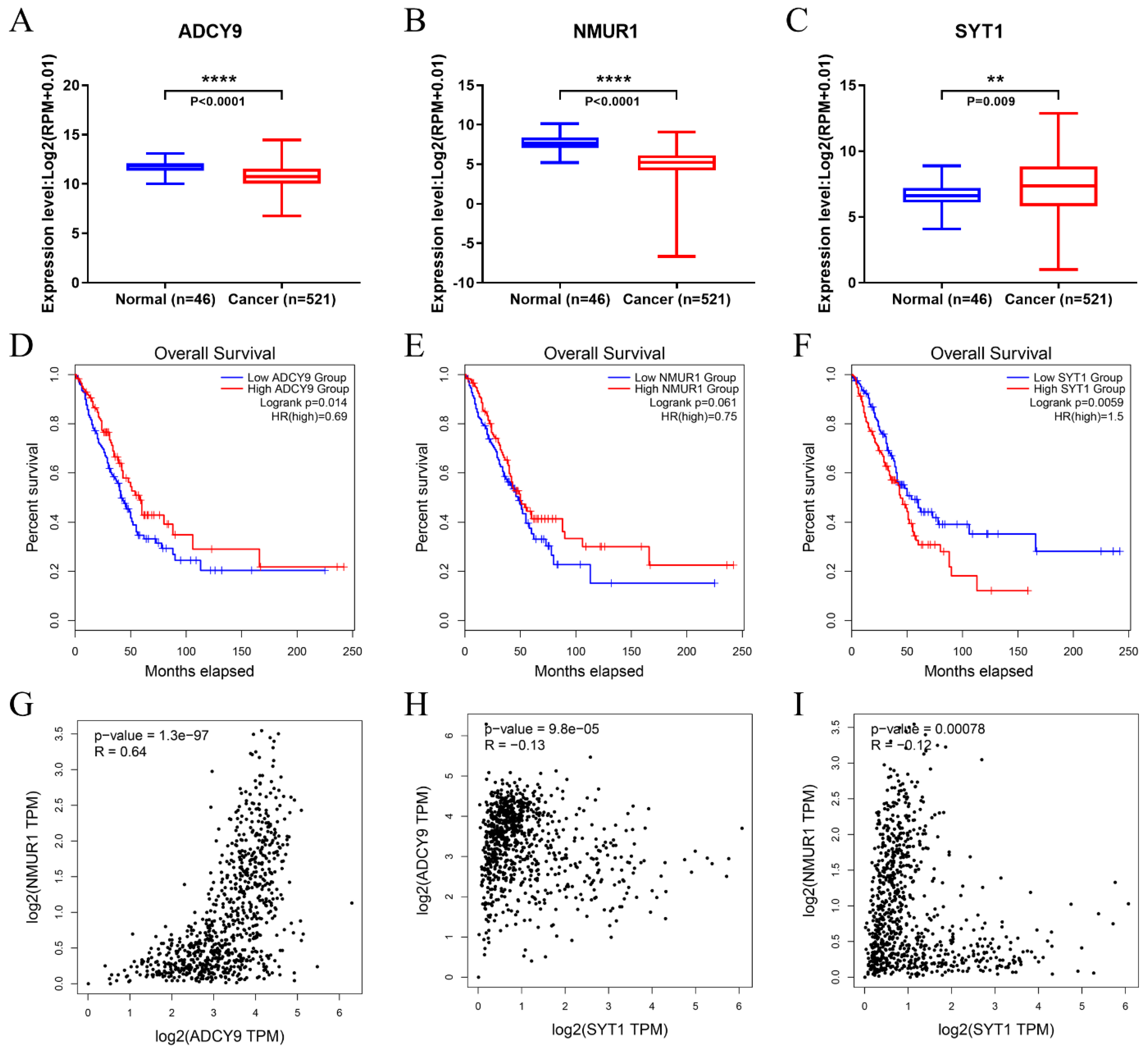
3.6. Identification of Hub Genes
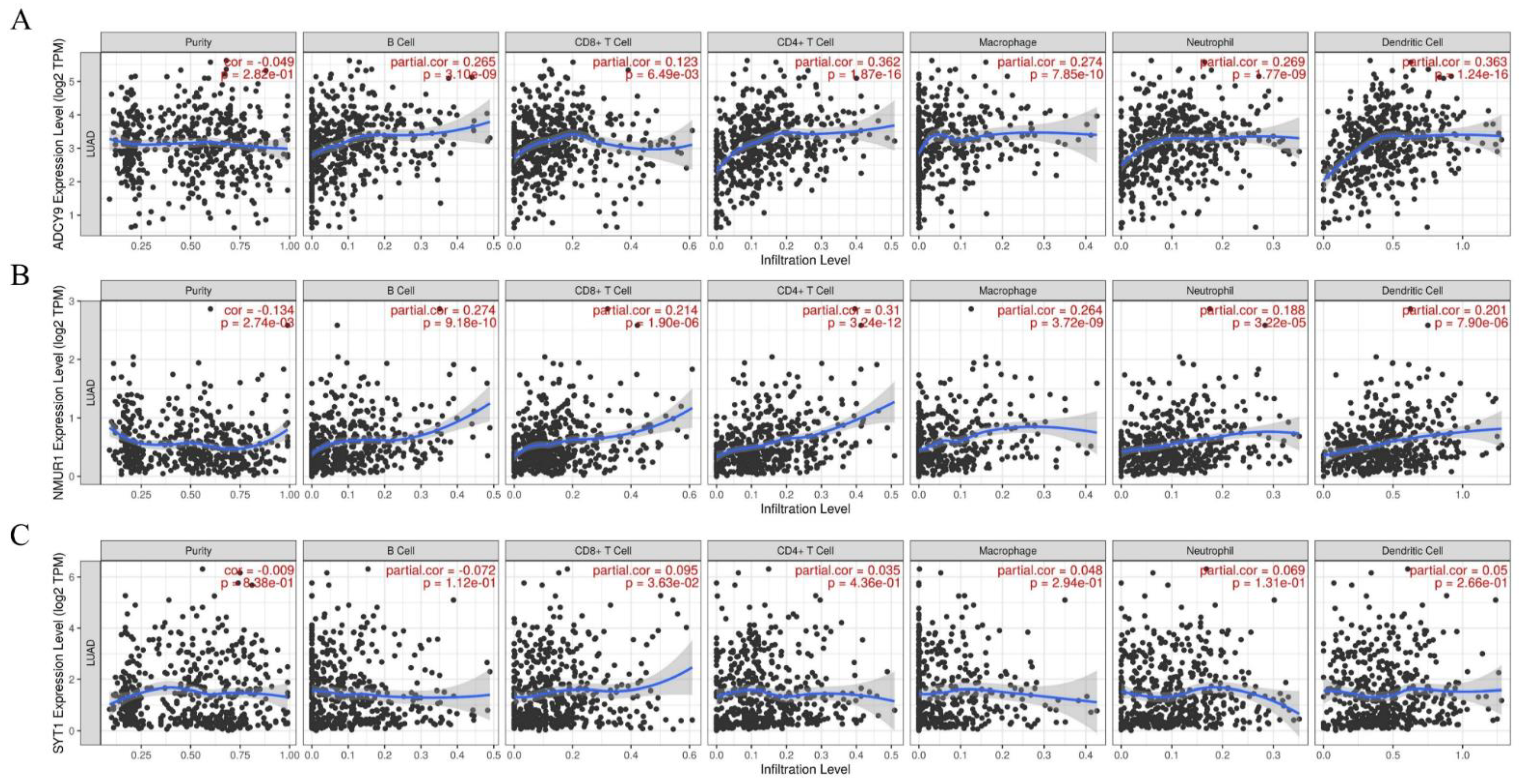
3.7. The Hub Genes Expression Was Correlated yo Immune Cell Infiltration in LUAD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.; Chirieac, L.R.; D’Amico, T.A.; DeCamp, M.M.; Dilling, T.J.; Dobelbower, M.; et al. Non-Small Cell Lung Cancer, Version 5. 2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 504–535. [Google Scholar]
- Meng, S.; Zhou, H.; Feng, Z.; Xu, Z.; Tang, Y.; Li, P.; Wu, M. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 2017, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zou, H.; Zhao, Y.; Chen, H.; Liu, T.; Wu, Z.; Yang, C.; Li, Q.; Li, Y. Tanshinone Inhibits NSCLC by Downregulating AURKA Through Let-7a-5p. Front. Genet. 2020, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, Y.; Liu, X.; Wu, Z.; Li, J.; Ma, Z. Roles of plant-derived bioactive compounds and related microRNAs in cancer therapy. Phytother. Res. 2021, 35, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Nan, A.; Chen, L.; Li, X.; Jia, Y.; Qiu, M.; Dai, X.; Zhou, H.; Zhu, J.; Zhang, H.; et al. Circular RNA circSATB2 promotes progression of non-small cell lung cancer cells. Mol. Cancer 2020, 19, 101. [Google Scholar] [CrossRef]
- Chen, X.; Mao, R.; Su, W.; Yang, X.; Geng, Q.; Guo, C.; Wang, Z.; Wang, J.; Kresty, L.A.; Beer, D.G.; et al. Circular RNA circHIPK3 modulates autophagy via MIR124-3p-STAT3-PRKAA/AMPKalpha signaling in STK11 mutant lung cancer. Autophagy 2020, 16, 659–671. [Google Scholar] [CrossRef]
- Huang, W.; Yang, Y.; Wu, J.; Niu, Y.; Yao, Y.; Zhang, J.; Huang, X.; Liang, S.; Chen, R.; Chen, S.; et al. Circular RNA cESRP1 sensitises small cell lung cancer cells to chemotherapy by sponging miR-93-5p to inhibit TGF-β signalling. Cell Death Differ. 2020, 27, 1709–1727. [Google Scholar] [CrossRef] [Green Version]
- Harrison, E.B.; Porrello, A.; Bowman, B.M.; Belanger, A.R.; Yacovone, G.; Azam, S.H.; Windham, I.A.; Ghosh, S.K.; Wang, M.; Mckenzie, N.; et al. A Circle RNA Regulatory Axis Promotes Lung Squamous Metastasis via CDR1-Mediated Regulation of Golgi Trafficking. Cancer Res. 2020, 80, 4972–4985. [Google Scholar] [CrossRef]
- Luo, Y.H.; Yang, Y.P.; Chien, C.S.; Yarmishyn, A.A.; Ishola, A.A.; Chien, Y.; Chen, Y.M.; Huang, T.W.; Lee, K.Y.; Huang, W.C.; et al. Plasma Level of Circular RNA hsa_circ_0000190 Correlates with Tumor Progression and Poor Treatment Response in Advanced Lung Cancers. Cancers 2020, 12, 1740. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, S.; Meng, N.; He, Y.; Lu, R.; Yan, G.R. ncRNA-Encoded Peptides or Proteins and Cancer. Mol. Ther. 2019, 27, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Salzman, J. Circular RNA Expression: Its Potential Regulation and Function. Trends Genet. 2016, 32, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Wang, Y.; Fan, Y.; Fang, N.; Wang, T.; Xu, T.; Shu, Y. CircRNAs in cancer metabolism: A review. J. Hematol. Oncol. 2019, 12, 90. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Huang, M.; Xing, L.; Yang, R.; Wang, X.; Jiang, R.; Zhang, L.; Chen, J. The circRNA circSEPT9 mediated by E2F1 and EIF4A3 facilitates the carcinogenesis and development of triple-negative breast cancer. Mol. Cancer 2020, 19, 73. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Hu, A.; Li, D.; Wang, J.; Guo, Y.; Liu, Y.; Li, H.; Chen, Y.; Wang, X.; Huang, K.; et al. Circ-HuR suppresses HuR expression and gastric cancer progression by inhibiting CNBP transactivation. Mol. Cancer 2019, 18, 158. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yu, J.; Huang, Z.; Fu, B.; Tao, Y.; Qi, X.; Mou, Y.; Hu, Y.; Wang, Y.; Cao, Y.; et al. Circular RNA hsa_circ_0000326 acts as a miR-338-3p sponge to facilitate lung adenocarcinoma progression. J. Exp. Clin. Cancer Res. 2020, 39, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, D.; Yang, S. MiR-490-3p Inhibits the Malignant Progression of Lung Adenocarcinoma. Cancer Manag. Res. 2020, 12, 10975–10984. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.N.; Ge, S.L.; Zhang, R.Q.; Huang, Y.L.; Liu, S.D.; Wu, K.M. MiR-490-3p inhibited the proliferation and metastasis of esophageal squamous cell carcinoma by targeting HMGA2. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8298–8305. [Google Scholar] [PubMed]
- Takagawa, Y.; Gen, Y.; Muramatsu, T.; Tanimoto, K.; Inoue, J.; Harada, H.; Inazawa, J. miR-1293, a Candidate for miRNA-Based Cancer Therapeutics, Simultaneously Targets BRD4 and the DNA Repair Pathway. Mol. Ther. 2020, 28, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Gordon, S.L.; Melland, H.; Bumbak, F.; Scott, D.J.; Jiang, T.J.; Owen, D.; Turner, B.J.; Boyd, S.G.; Rossi, M.; et al. SYT1-associated neurodevelopmental disorder: A case series. Brain 2018, 141, 2576–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Zeng, X.; Wu, B.; Zhao, J.; Pan, Y. RNA-Seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with radiotherapy response of nasopharyngeal carcinoma and prognosis of head and neck cancer. Cancer Biol. Ther. 2020, 21, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Hao, L.; Yang, H.; Chen, J.; Liu, J. miRNA-34a suppresses colon carcinoma proliferation and induces cell apoptosis by targeting SYT1. Int. J. Clin. Exp. Pathol. 2019, 12, 2887–2897. [Google Scholar] [PubMed]
- Zhang, G.; Xi, M.; Li, Y.; Wang, L.; Gao, L.; Zhang, L.; Yang, Z.; Shi, H. The ADCY9 genetic variants are associated with glioma susceptibility and patient prognosis. Genomics 2021, 113, 706–716. [Google Scholar] [CrossRef]
- Hopewell, J.C.; Ibrahim, M.; Hill, M.; Shaw, P.M.; Braunwald, E.; Blaustein, R.O.; Bowman, L.; Landray, M.J.; Sabatine, M.S.; Collins, R.; et al. Impact of ADCY9 Genotype on Response to Anacetrapib. Circulation 2019, 140, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, Y.L.; Liu, S.; Zhang, P.P.; Chen, Z.; Liu, M.; Tang, H. miR-181b promotes cell proliferation and reduces apoptosis by repressing the expression of adenylyl cyclase 9 (AC9) in cervical cancer cells. FEBS Lett. 2014, 588, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Misawa, K.; Mima, M.; Satoshi, Y.; Misawa, Y.; Imai, A.; Mochizuki, D.; Nakagawa, T.; Kurokawa, T.; Oguro, M.; Ishikawa, R.; et al. Neuropeptide receptor genes GHSR and NMUR1 are candidate epigenetic biomarkers and predictors for surgically treated patients with oropharyngeal cancer. Sci. Rep. 2020, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Zhang, J.; Ma, X.; Tan, J.; Chen, L.; Liu, S.; Xin, Y.; Zhuang, L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed. Pharmacother. 2020, 131, 110678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Source | Platform | First Author | Year | Region | Sample Size (T/N) | Number of circRNAs | Title |
|---|---|---|---|---|---|---|---|
| GSE101684 | GPL21825 | Ming Xu | 2019 | China | 4/4 | 9114 | CircRNA expression profiles in early stages lung adenocarcinoma. |
| GSE112214 | GPL19978 | Yeping Dong | 2019 | China | 3/3 | 4407 | Genome-wide analysis of circRNA expression profile in non-small cell lung cancer (NSCLC) tissues and matched adjacent normal tissues. |
| GSE158695 | GPL19978 | Botai Li | 2020 | China | 3/3 | 1941 | circRNA expression data from human non-small cell lung cancer tissues and the correponding non-cancerous tissues. |
| GSE101586 | GPL19978 | Mantang Qiu | 2017 | China | 5/5 | 4425 | Profiling circular RNA expression in lung cancer. |
| CircRNA | Alias | Position | Best Transcript | Gene Symbol | Length | Regulation |
|---|---|---|---|---|---|---|
| hsa_circRNA_103415 | hsa_circ_0008234 | chr3: 71090478-71102924 strand: − | NM_001244808 | FOXP1 | 587 | Up |
| hsa_circRNA_101066 | hsa_circ_0026337 | chr12: 52180325-52188425 strand: + | NM_014191 | SCN8A | 853 | Up |
| hsa_circRNA_104513 | hsa_circ_0007518 | chr7: 140402665-140404763 strand: + | NM_004546 | NDUFB2 | 249 | Up |
| hsa_circRNA_103134 | hsa_circ_0061749 | chr21: 40648098-40649277 strand: − | NM_033656 | BRWD1 | 142 | Up |
| hsa_circRNA_100395 | hsa_circ_0015278 | chr1: 173726114-173744981 strand: + | NM_014458 | KLHL20 | 671 | Up |
| hsa_circRNA_101213 | hsa_circ_0029426 | chr12: 131357380-131357465 strand: + | NM_006325 | RAN | 85 | Up |
| hsa_circRNA_102046 | hsa_circ_0043256 | chr17: 35604934-35609962 strand: − | NM_198839 | ACACA | 483 | Up |
| hsa_circRNA_102442 | hsa_circ_0049271 | chr19: 10610070-10610756 strand: − | NM_203500 | KEAP1 | 686 | Up |
| hsa_circRNA_002178 | hsa_circ_0000519 | chr14: 20811436-20811534 strand: − | NR_002312 | RPPH1 | 98 | Down |
| hsa_circRNA_103123 | hsa_circ_0002360 | chr21: 36206706-36231875 strand: − | NM_001001890 | RUNX1 | 297 | Down |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Zou, H. Identification of the circRNA-miRNA-mRNA Prognostic Regulatory Network in Lung Adenocarcinoma. Genes 2022, 13, 885. https://doi.org/10.3390/genes13050885
Ma Y, Zou H. Identification of the circRNA-miRNA-mRNA Prognostic Regulatory Network in Lung Adenocarcinoma. Genes. 2022; 13(5):885. https://doi.org/10.3390/genes13050885
Chicago/Turabian StyleMa, Yan, and Heng Zou. 2022. "Identification of the circRNA-miRNA-mRNA Prognostic Regulatory Network in Lung Adenocarcinoma" Genes 13, no. 5: 885. https://doi.org/10.3390/genes13050885
APA StyleMa, Y., & Zou, H. (2022). Identification of the circRNA-miRNA-mRNA Prognostic Regulatory Network in Lung Adenocarcinoma. Genes, 13(5), 885. https://doi.org/10.3390/genes13050885