
Novel Loss-of-Function Variants in CHD2 Cause Childhood-Onset Epileptic Encephalopathy in Chinese Patients

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Genetic Analysis
2.3. Deposition of Sequences
2.4. Molecular Analysis
3. Results
3.1. Clinical Features
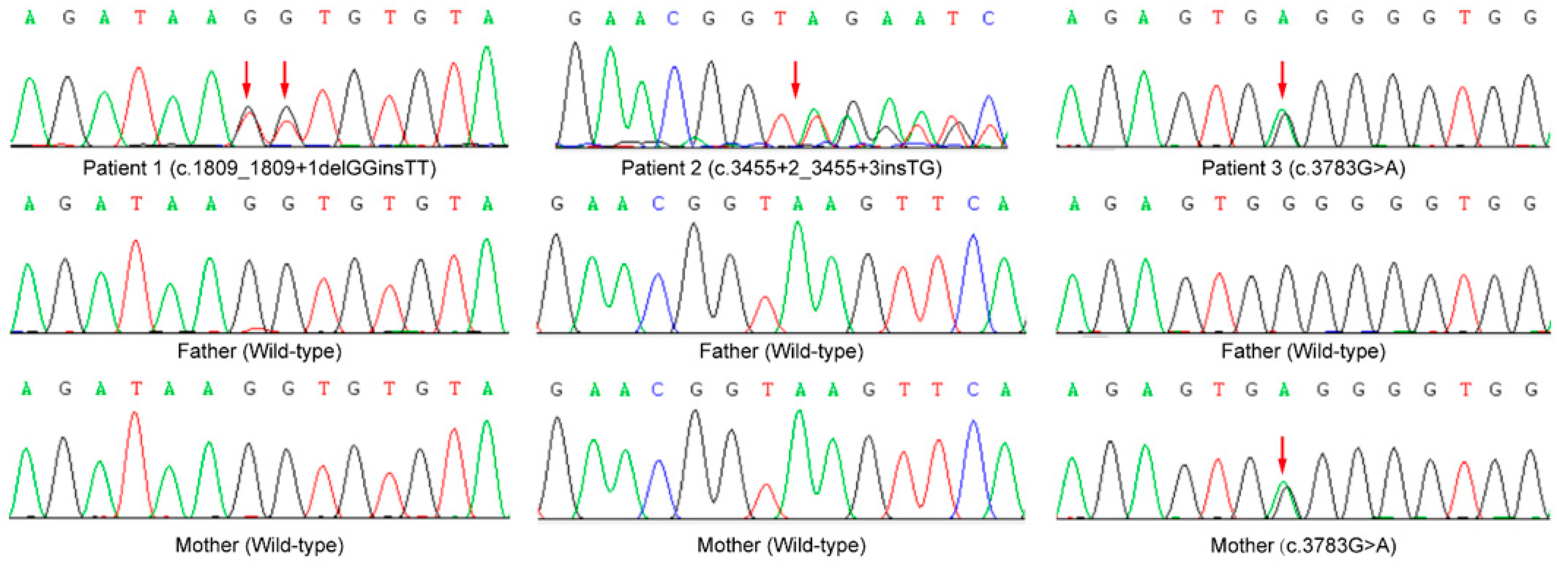
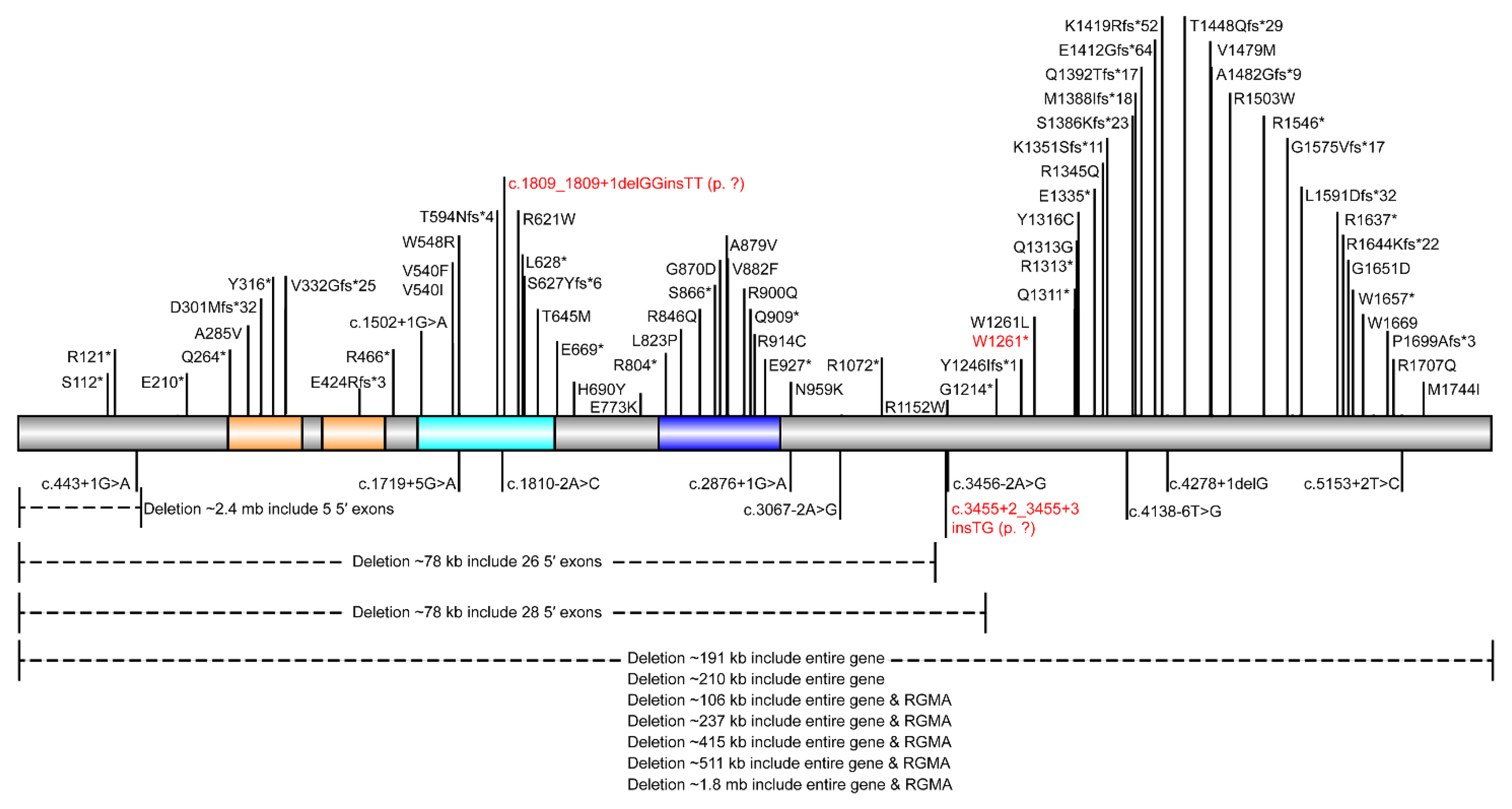
3.2. Sequencing Results
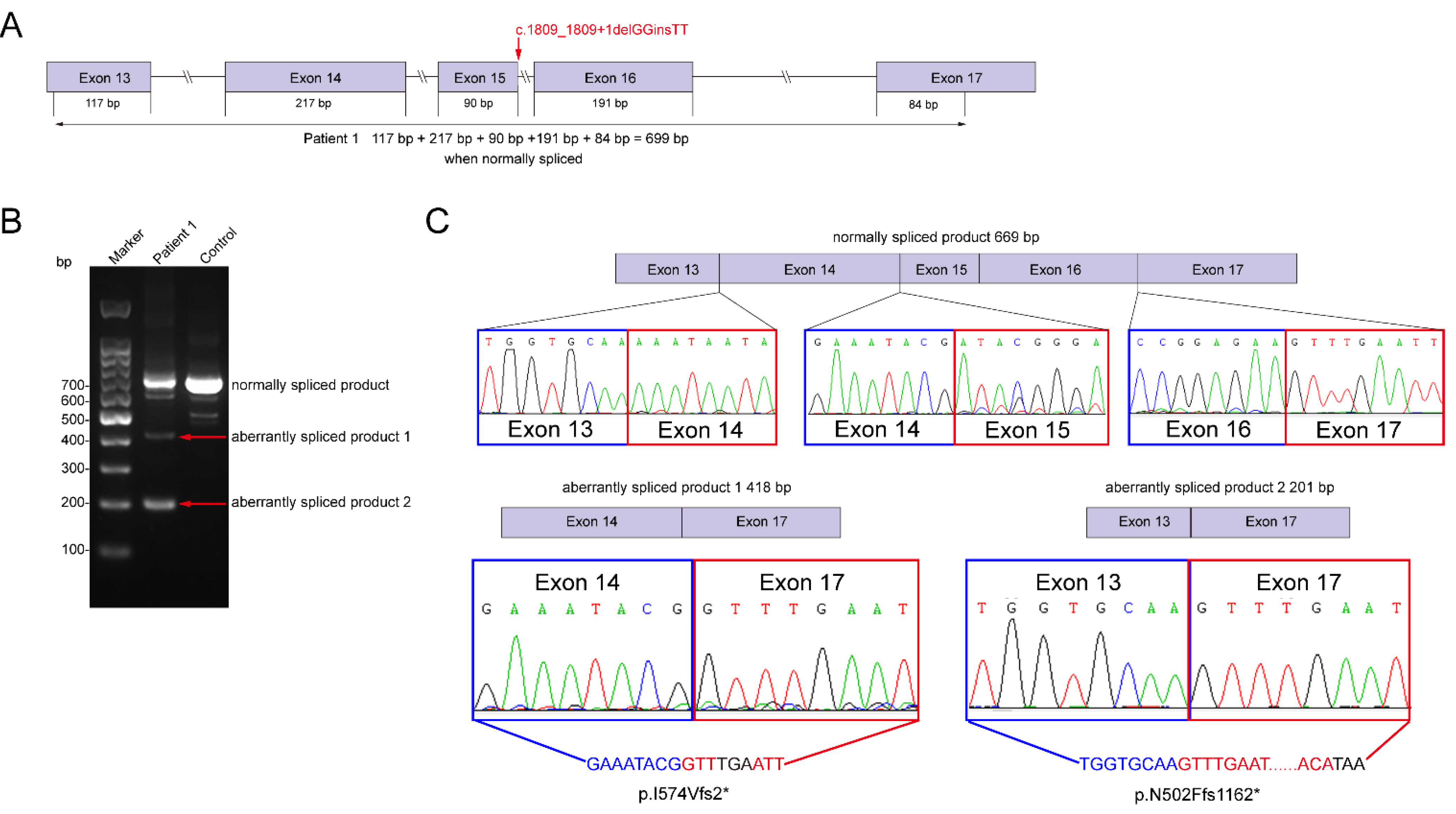
3.3. Molecular Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Statement
References
- Covanis, A. Epileptic Encephalopathies (Including Severe Epilepsy Syndromes). Epilepsia 2012, 53 (Suppl. S4), 114–126. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.D.; Brooks-Kayal, A.R.; Porter, B.E. Seizures and Antiepileptic Drugs: Does Exposure Alter Normal Brain Development? Epilepsia 2006, 47, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Kamien, B.A.; Cardamone, M.; Lawson, J.A.; Sachdev, R. A Genetic Diagnostic Approach to Infantile Epileptic Encephalopathies. J. Clin. Neurosci. 2012, 19, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Capelli, L.P.; Krepischi, A.C.; Gurgel-Giannetti, J.; Mendes, M.F.S.; Rodrigues, T.; Varela, M.C.; Koiffmann, C.P.; Rosenberg, C. Deletion of the RMGA and CHD2 Genes in a Child with Epilepsy and Mental Deficiency. Eur. J. Med. Genet. 2012, 55, 132–134. [Google Scholar] [CrossRef]
- Petersen, A.K.; Streff, H.; Tokita, M.; Bostwick, B.L. The First Reported Case of an Inherited Pathogenic CHD2 Variant in a Clinically Affected Mother and Daughter. Am. J. Med. Genet. Part A 2018, 176, 1667–1669. [Google Scholar] [CrossRef]
- Veredice, C.; Bianco, F.; Contaldo, I.; Orteschi, D.; Stefanini, M.C.; Battaglia, D.I.; Lettori, D.; Guzzetta, F.; Zollino, M. Early Onset Myoclonic Epilepsy and 15q26 Microdeletion: Observation of the First Case. Epilepsia 2009, 50, 1810–1815. [Google Scholar] [CrossRef]
- Lemke, J.R. Predicting Incidences of Neurodevelopmental Disorders. Brain 2020, 143, 1046–1048. [Google Scholar] [CrossRef]
- Chénier, S.; Yoon, G.; Argiropoulos, B.; Lauzon, J.; Laframboise, R.; Ahn, J.W.; Ogilvie, C.M.; Lionel, A.C.; Marshall, C.R.; Vaags, A.K.; et al. CHD2 Haploinsufficiency Is Associated with Developmental Delay, Intellectual Disability, Epilepsy and Neurobehavioural Problems. J. Neurodev. Disord. 2014, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Guo, J.; Hao, C.; Guo, R.; Hu, X.; Chen, Y.; Yang, W.; Li, W.; Feng, Y. Whole-Exome Sequencing Reveals Two De Novo Variants in the RBM20 Gene in Two Chinese Patients with Left Ventricular Non-Compaction Cardiomyopathy. Pediatr. Investig. 2020, 4, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the Molecular and Phenotypic Impact of Amino Acid Variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Brunak, S.; Engelbrecht, J.; Knudsen, S. Prediction of Human mRNA Donor and Acceptor Sites from the DNA Sequence. J. Mol. Biol. 1991, 220, 49–65. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ferlaino, M.; Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. An Integrative Approach to Predicting the Functional Effects of Small Indels in Non-Coding Regions of the Human Genome. BMC Bioinform. 2017, 18, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Shen, X.; Fang, F.; Ding, C.-H.; Zhang, H.; Cao, Z.-H.; An, D.-Y. Phenotype-Driven Virtual Panel Is an Effective Method to Analyze WES Data of Neurological Disease. Front. Pharmacol. 2019, 9, 1529. [Google Scholar] [CrossRef]
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised Terminology and Concepts for Organization of Seizures and Epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef]
- Wang, Y.; Du, X.; Bin, R.; Yu, S.; Xia, Z.; Zheng, G.; Zhong, J.; Zhang, Y.; Jiang, Y.-H.; Wang, Y. Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. Sci. Rep. 2017, 7, 40319. [Google Scholar] [CrossRef] [Green Version]
- Galizia, E.C.; Myers, C.T.; Leu, C.; de Kovel, C.G.F.; Afrikanova, T.; Cordero-Maldonado, M.L.; Martins, T.G.; Jacmin, M.; Drury, S.; Chinthapalli, V.K.; et al. Chd2 Variants Are a Risk Factor for Photosensitivity in Epilepsy. Brain 2015, 138, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, J. Chd2-Related Epilepsy: Novel Mutations and New Phenotypes. Dev. Med. Child Neurol. 2020, 62, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Chen, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Neurological Diseases with Autism Spectrum Disorder: Role of ASD Risk Genes. Front. Neurosci. 2019, 13, 349. [Google Scholar] [CrossRef] [PubMed]
- Suls, A.; Jaehn, J.A.; Kecskés, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.; Siekierska, A.; Djémié, T.; Afrikanova, T.; Gormley, P.; et al. De Novo Loss-of-Function Mutations in CHD2 Cause a Fever-Sensitive Myoclonic Epileptic Encephalopathy Sharing Features with Dravet Syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.H.; Zhang, L.M.; Carvill, G.L.; Archer, J.S.; Heavin, S.B.; Mandelstam, S.; Craiu, D.; Berkovic, S.F.; Gill, D.; Mefford, H.C.; et al. CHD2 Myoclonic Encephalopathy Is Frequently Associated with Self-Induced Seizures. Neurology 2015, 84, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Poisson, A.; Chatron, N.; Labalme, A.; Fourneret, P.; Ville, D.; Mathieu, M.L.; Sanlaville, D.; Demily, C.; Lesca, G. Chromatin Remodeling Dysfunction Extends the Etiological Spectrum of Schizophrenia: A Case Report. BMC Med. Genet. 2020, 21, 10–16. [Google Scholar] [CrossRef]
- Wang, J.; Wen, Y.; Zhang, Q.; Yu, S.; Chen, Y.; Wu, X.; Zhang, Y.; Bao, X. Gene Mutational Analysis in a Cohort of Chinese Children with Unexplained Epilepsy: Identification of a New KCND3 Phenotype and Novel Genes Causing Dravet Syndrome. Seizure 2019, 66, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Trivisano, M.; Striano, P.; Sartorelli, J.; Giordano, L.; Traverso, M.; Accorsi, P.; Cappelletti, S.; Claps, D.J.; Vigevano, F.; Zara, F.; et al. CHD2 Mutations Are a Rare Cause of Generalized Epilepsy with Myoclonic–Atonic Seizures. Epilepsy Behav. 2015, 51, 53–56. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Proband | 1 | 2 | 3 | Patients Reported Previously (n = 65) |
|---|---|---|---|---|
| Gender | M | F | M | M (38)/F (26)/NK (1) |
| Variants | c.1809_1809+1delGGinsTT (p. ?) 15:93498742:93498743:GG:TT | c.3455+2_3455+3insTG (p. ?) 15:93534749:93534749:T:TTG | c.3783G>A (p.W1261*) 15:93540531:93540531:G:A | Nonsense (17)/Frameshift (18)/Splice (6)/Missense (15)/Deletion (9) |
| Inheritance | De novo | De novo | Maternal | De novo (51)/Paternal (3)/Maternal (1)/NK (10) |
| Family History of Epilepsy | - | + | - | Yes (8)/No (23)/NK (34) |
| ACMG classification | P (PVS1+PS2+PM2) | LP (PS2+PM2+PP3) | LP (PVS1+PM2) | P (42)/LP (21)/VUS (2) |
| Onset ages | 2y3m | 2y11m | 6y4m | 6m-10y5m |
| Brain Imaging (MRI) | - | Enlarged subarachnoid space in the frontal and temporal region | Space-occupying lesion of the right thalamus | Normal (25)/Abnormal (10)/NK (30) |
| Seizure Types (patients can have multiple seizure types) | Patients reported with different seizure types (n = 59) | |||
| -Seizures | + | + | Solo (1)/Combined (8) | |
| -Epileptic spasm | + | Solo (0)/Combined (2) | ||
| -Status epilepticus | Solo (0)/Combined (5) | |||
| -Atonic seizures | Solo (0)/Combined (11) | |||
| -Clonic seizures | Solo (0)/Combined (2) | |||
| -Tonic seizures | Solo (1)/Combined (7) | |||
| -Generalized tonic seizures | Solo (0)/Combined (2) | |||
| -Myoclonic seizures | + | Solo (2)/Combined (28) | ||
| -Focal myoclonic seizures | Solo (0)/Combined (1) | |||
| -Generalized myoclonic seizures | Solo (1)/Combined (1) | |||
| -Focal seizures | + | Solo (1)/Combined (5) | ||
| -Focal impaired awareness seizures | Solo (0)/Combined (1) | |||
| -Absence seizures | Solo (4)/Combined (11) | |||
| -Absence seizures (Atypical) | Solo (0)/Combined (10) | |||
| -Absence seizures (with Eyelid myoclonia) | Solo (0)/Combined (5) | |||
| -Generalized seizures | + | Solo (1)/Combined (2) | ||
| -Generalized myoclonic-absence seizures | Solo (0)/Combined (3) | |||
| -Generalized myoclonic-atonic seizures | Solo (0)/Combined (5) | |||
| -Generalized myoclonic-atonic-absence seizures | Solo (0)/Combined (3) | |||
| -Generalized myoclonic-clonic seizures | Solo (0)/Combined (1) | |||
| -Generalized tonic-clonic seizures | + | + | Solo (6)/Combined (32) | |
| -Febrile seizures | + | + | + | Solo (0)/Combined (8) |
| Fever Sensitivity of Seizure Onset | + | + | + | Yes (14)/No (14)/NK (31) |
| Developmental Delay Before Epilepsy | + | - | + | Yes (17)/No (8)/NK (34) |
| EEG/VEEG | Multifocal epileptic discharges at 3y7m; High-amplitude sharp and slow wave, slow spike wave, slow polyspike wave at 5y3m; Generalized spike waves, slow spike wave, slow polyspike wave at 7y3m | Focal epileptic discharges, 2–3Hz polyspike wave at 5y | Diffuse polyspike wave, slow spike wave at 6y5m | Abnormal(33)/Normal(0)/NK(26) |
| Photosensitivity | - | NK | NK | Yes (16)/No (2)/NK (41) |
| Treatment | Patients reported with detailed medication history (n = 37) | |||
| -Bromide | Solo (0)/Combined (2) | |||
| -Clobazam | Solo (0)/Combined (7) | |||
| -Clonazepam | + | Solo (0)/Combined (10) | ||
| -Ethosuximide | Solo (0)/Combined (4) | |||
| -Lamotrigine | + | Solo (1)/Combined (14) | ||
| -Levetiracetam | + | Solo (1)/Combined (20) | ||
| -Oxcarbazepine | + | Solo (0)/Combined (4) | ||
| -Phenobarbital | Solo (0)/Combined (3) | |||
| -Phenytoin | Solo (0)/Combined (1) | |||
| -Rufinamide | Solo (0)/Combined (2) | |||
| -Topiramate | + | Solo (0)/Combined (10) | ||
| -Valproic acid | + | + | Solo (4)/Combined (34) | |
| -Vigabatrin | Solo (0)/Combined (1) | |||
| -Zonisamide | Solo (0)/Combined (4) | |||
| -Ketogenic diet | + | Solo (0)/Combined (5) | ||
| Seizure Remission (age) | Yes (6y7m) | Yes (5y1m) | Yes (8y4m) | Yes (18)/Partially (2)/No (12)/NK (5) Effective medication or combination: Valproic acid + Levetiracetam (4); Valproic acid + Lamotrigine (2); Valproic acid (2); Levetiracetam (1); Lamotrigine (1); Valproic acid + Clonazepam (1); Valproic acid + Ethosuximide + Lamotrigine (1); Valproic acid + Clobazam + Lamotrigine (1); Valproic acid + Clobazam + Rufinamide (1); Valproic acid + Levetiracetam + Topiramate (1); Valproic acid + Levetiracetam + Topiramate + Clobazam (1); Valproic acid + Levetiracetam + Topiramate + Lamotrigine + Zonisamide (1); Valproic acid + Clonazepam + Lamotrigine + Levetiracetam + Oxcarbazepine + Phenobarbital + Topiramate (1) |
| Development (Psychomotor and Cognition) | Patients reported previously (n = 65) | |||
| -ID | + | + | n = 27 | |
| -Delayed psychomotor development | Delayed language and motor development | Moderate developmental retardation | n = 46 | |
| -Psychomotor regression | Intellectual regression | n = 17 | ||
| Behavior | ||||
| -ASD | n = 23 | |||
| -ADHD | n = 7 | |||
| -Aggressive behavior | n = 17 | |||
| -Psychotic features | n = 9 | |||
| -Bruxism | n = 2 | |||
| -Stereotypic movements | n = 4 | |||
| -Limited social skills | n = 3 | |||
| -Short attention span | n = 2 | |||
| Dysmorphic features | Microcephaly | Slender fingers | Normal (52)/Abnormal (13) | |
| Other | Hypotonia in infancy | Hypotonia (7), scoliosis (5), feeding difficulties (2), visual disability (3), short stature (4), gait (7), ataxia (7) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Cui, D.; Ding, C.; Chen, C.; Wang, X.; Fang, F.; Jin, H.; Ren, X. Novel Loss-of-Function Variants in CHD2 Cause Childhood-Onset Epileptic Encephalopathy in Chinese Patients. Genes 2022, 13, 908. https://doi.org/10.3390/genes13050908
Wang X, Cui D, Ding C, Chen C, Wang X, Fang F, Jin H, Ren X. Novel Loss-of-Function Variants in CHD2 Cause Childhood-Onset Epileptic Encephalopathy in Chinese Patients. Genes. 2022; 13(5):908. https://doi.org/10.3390/genes13050908
Chicago/Turabian StyleWang, Xu, Di Cui, Changhong Ding, Chunhong Chen, Xiaohui Wang, Fang Fang, Hong Jin, and Xiaotun Ren. 2022. "Novel Loss-of-Function Variants in CHD2 Cause Childhood-Onset Epileptic Encephalopathy in Chinese Patients" Genes 13, no. 5: 908. https://doi.org/10.3390/genes13050908
APA StyleWang, X., Cui, D., Ding, C., Chen, C., Wang, X., Fang, F., Jin, H., & Ren, X. (2022). Novel Loss-of-Function Variants in CHD2 Cause Childhood-Onset Epileptic Encephalopathy in Chinese Patients. Genes, 13(5), 908. https://doi.org/10.3390/genes13050908