
Biallelic Optic Atrophy 1 (OPA1) Related Disorder—Case Report and Literature Review


Abstract
:1. Introduction
2. Materials and Methods
3. Results
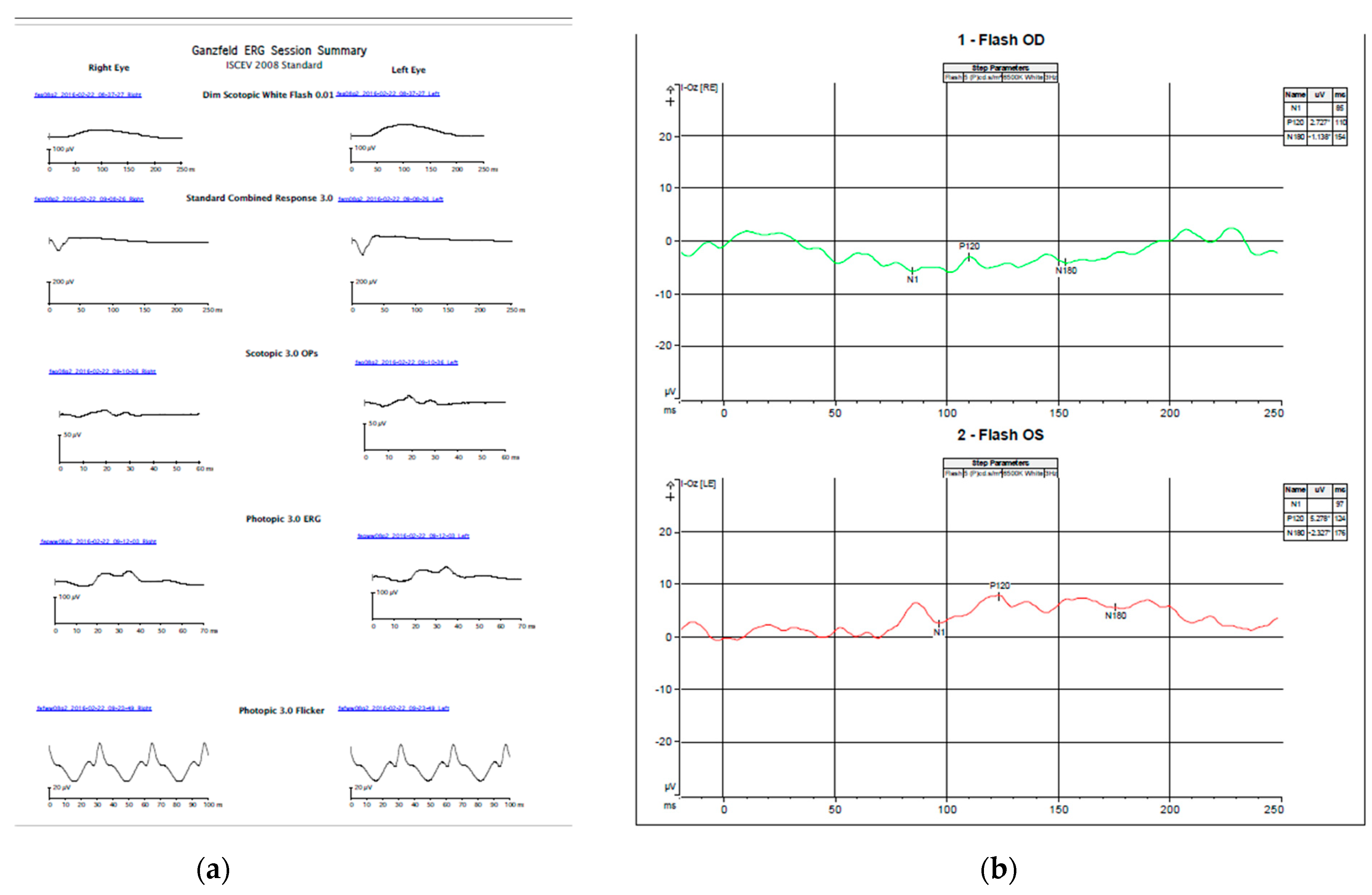
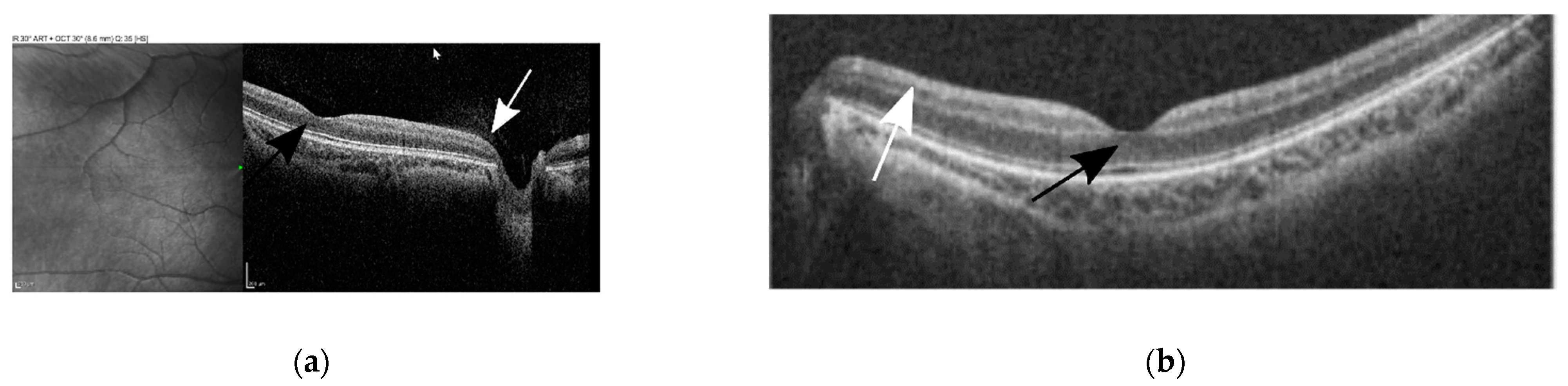
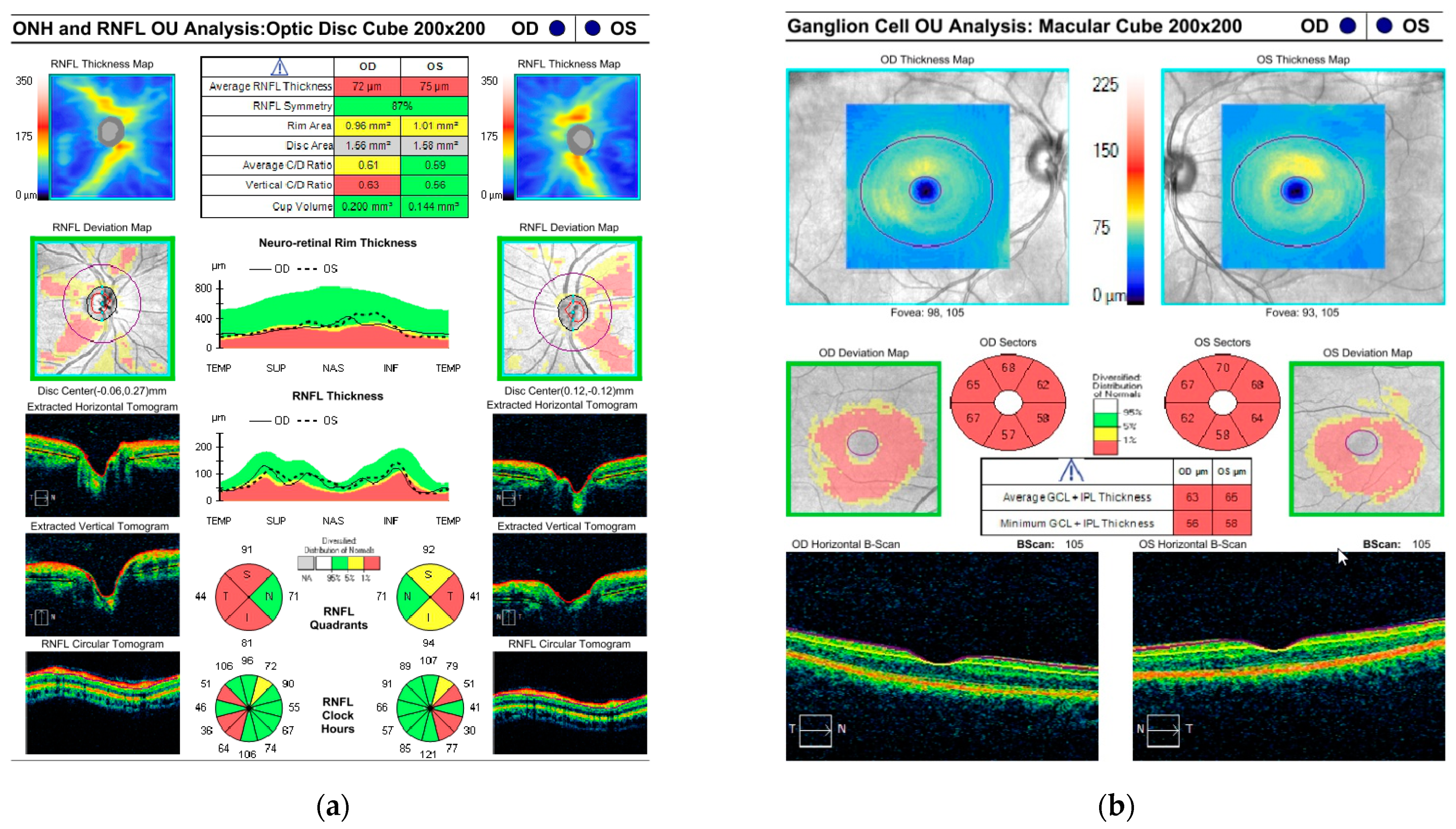
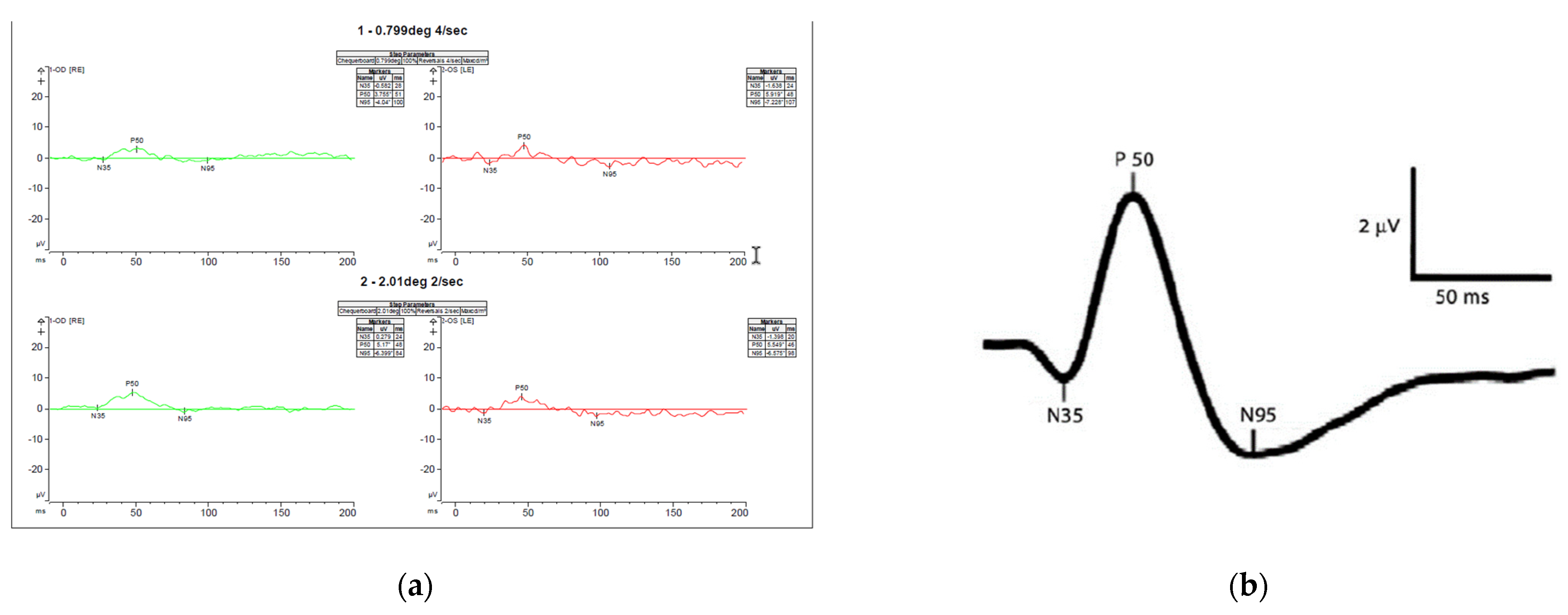
3.1. Case Description
3.2. Genetic Testing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Age of Onset | Gender | Mutation/Amino Acid Change | Visual Acuity | Optic Atrophy | Ataxia | Peripheral Neuropathy | Seizures | VEP | Brain MRI | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | ||||||||||
| The current report | Birth | M | c.1311A>G/ p.lIle437Met | c.2287del/ p.Ser763Valfs*15 | 0.05/0.1 | Yes | Yes | No | status epilepticus and intractable seizures | Low amplitude | Multiple metabolic strokes in his left thalamus, left occipital lobe, and left frontal lobe |
| Bonneau et al. [23] | 42 mo | F | c.1146A>G/ p.Ile382Met | c.2470C>T/ p.Arg824 * | 0.01/0.01 | Yes | Yes | Yes | Not reported | Abnormal | Normal |
| Bonneau et al. [23] | 12 mo | F | c.1204G>A/ p.Val402Met | c.2708-2711del4/ p.Val903Glyfs * | unknown | Yes | Yes | Yes | Not reported | Abnormal | Cerebellar atrophy |
| Bonneau et al. [23] | 18 mo | M | c.1146A>G/ p.Ile382Met | c.1669C>T/ p.Arg557 * | 0.01/0.01 | Yes | Yes | Yes | Not reported | Abnormal | Cerebellar atrophy |
| Bonneau et al. [23] | 3 y | M | c.1146A>G/ p.Ile382Met | c.1459G>A/ p.Glu487Lys | Blind | Yes | Yes | Yes | Not reported | Abnormal | Cerebellar atrophy. Hypoplasia of the optic chiasm and optic nerves |
| Schaaf et al. [25] | 12 mo | M | c.1146A>G/ p.Ile382Met | c.2708-2711del4/ p.Val903Glyfs * | Reduced severely | Severe, diffuse | Yes | Yes | No | unknown | Periventricualr leucomalacia |
| Schaaf et al. [25] | 6 mo | F | c.1146A>G/ p.Ile382Met | c.2708-2711del4/ p.Val903Glyfs * | unknown | Severe, diffuse | Yes | Yes | No | unknown | Not performed |
| Pesch et al. [26] | Childhood | F | c.808G>A/ p.Glu270Lys | c.868C>T/ p.Arg290Trp | 0.2/0.2 | Diffuse pallor | Not reported | Yes | No | Abnormal—delayed latencies | unknown |
| Lee et al. [27] | 12 mo | M | c.2714G>A/ p.Arg905Gln | c.1857-1858delinsT/ p.Leu620fs*13 | Blind. Congenital cataract | Diffuse pallor | Yes | Yes | No | Absent | Normal |
| Yu-Wai-Man et al. [6] | unknown | M | c.768C>G/ p.Ser256Arg | c.854A>G/ p.Gln285Arg | Unk | Yes | Yes | Yes | Not reported | unknown | unknown |
| Yu-Wai-Man et al. [6] | unknown | F | c.768C>G/ p.Ser256Arg | c.854A>G/ p.Gln285Arg | Unk | Yes | Yes | Yes | Not reported | unknown | unknown |
| Carelli et al. [28] | 1 y | M | c.1311A>G/ p.lIle437Met | c.1705+1G>T | LP | Yes | Yes | Yes | No | Delayed | Normal |
| Bonifert et al. [12] | 2 y | M | c.1311A>G/ p.lIle437Met | c.610+364G>A | 0.02 | Yes | Yes | Yes | No | unknown | Cerebellar atrophy and thinning of the cervical spinal cord |
| Bonifert et al. [12] | 2 y | M | c.1311A>G/ p.lIle437Met | c.610+364G>A | 0.02 | Yes | Yes | Yes | No | unknown | Cerebellar atrophy and thinning of the cervical spinal cord |
| Bonifert et al. [12] | 2 y | F | c.1311A>G/ p.lIle437Met | c.610+364G>A | 0.02 | Yes | Yes | Yes | No | unknown | Cerebellar atrophy and thinning of the cervical spinal cord |
| Bonifert et al. [12] | 1 y | F | c.1311A>G/ p.lIle437Met | c.1316_1317insA/ p.N440Kfs*14 | unknown | Yes | Yes | Yes | No | unknown | Unknown |
| Nasca et al. [15] | 1 mo | M | c.1311A>G/ p.lIle437Met | c.190_194del/ p.Ser64Asnfs*7 | unknown | Yes | Yes | Yes | Present | unknown | Cerebellar atrophy, edema in the occipito-parietal cortical areas |
| Nasca et al. [15] | 4-6 y | F | c.1311A>G/ p.lIle437Met | c.2962G>T/ p.Val988Phe | unknown | Yes | Yes | Yes | No | Abnormal - optic nerve dysfunction | Optic nerves and chiasm atrophy |
| Nasca et al. [15] | 4 y | F | c.1180G>A/ p.Ala394Thr | c.1180G>A/ p.Ala394Thr | unknown | Unk | Yes | Yes | No | Abnormal - optic nerve dysfunction | Abnormal hyperintensities of the periventricular and centrum semiovale white matter areas, mild global cerebellar atrophy |
| Zerem et al. [14] | 18 mo | F | c.1146A>G/ p.Ile382Met | c.1963_1964dupAT/ p.Lys656fs | 0.008/0.004 | Yes | Yes | Yes | Not reported | Low amplitude responses | Atrophy of the optic nerves, mild central and deep white matter multifocal T2 hyperintensities. Right parietal acute infarct. Metabolic stroke |
| Rubegni et al. [29] | 5 y | F | c.1180G>A/ p.Ala394Thr | c.1180G>A/ p.Ala394Thr | unknown | Optic disc pallor | Yes | Yes | No | unknown | Progressive cerebellar involvement accompanied by basal ganglia hyperintensities |
| Rubegni et al. [29] | 8 mo | M | c.2779-2A>C | c.2809C>T/ p.Arg937Cys | Blindness | Severe optic atrophy | Yes | Yes | No | Increase latency and reduced amplitude | Progressive cerebellar involvement accompanied by basal ganglia hyperintensities |
3.3. Parental Examination
3.4. Management and Disease Progression
4. Discussion and Literature Review
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2021, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrick, Y.W.M.; Chinnery, P.F. Dominant optic atrophy: Novel OPA1 mutations and revised prevalence estimates. Ophthalmology 2013, 120, 1712–1712.e1. [Google Scholar]
- Carelli, V.; Ross, F.N.C.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Guillou, E.; Delettre, C.; Landes, T.; Laetitia, A.-P.; Emorinea, L.J.; Mils, V.; Daloyau, M.; Hamel, C.; Patrizia, A.B. Mitochondrial dynamics and disease, OPA1. Biochim. Et Biophys. Acta Mol. Cell Res. 2006, 1763, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- le Roux, B.; Lenaers, G.; Zanlonghi, X.; Patrizia, A.B.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 124. [Google Scholar] [CrossRef]
- Man, P.Y.W.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer, M.G.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar]
- Monaco, F.; Pirisi, A.; Sechi, G.P.; Mutani, R. Complicated optic atrophy (Behr’s disease) associated with epilepsy and amino acid imbalance. Eur. Neurol. 1979, 18, 101–105. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Anikster, Y.; Kleta, R.; Shaag, A.; Gahl, W.A.; Elpeleg, O. Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or costeff optic atrophy syndrome): Identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am. J. Hum. Genet. 2001, 69, 1218–1224. [Google Scholar] [CrossRef] [Green Version]
- Costeff, H.; Gadoth, N.; Apter, N.; Prialni, M.; Savir, H. A familial syndrome of infantile optic atrophy, movement disorder, and spastic paraplegia. Neurology 1989, 2, 595–597. [Google Scholar] [CrossRef]
- Costeff, H.; Elpeleg, O.; Apter, N.; Divry, P.; Gadoth, N. 3-Methylglutaconic aciduria in ‘optic atrophy plus’. Ann. Neurol. 1993, 33, 103–104. [Google Scholar] [CrossRef]
- Bonifert, T.; Karle, K.N.; Tonagel, F.; Batra, M.; Wilhelm, C.; Theurer, Y.; Schoenfeld, C.; Kluba, T.; Kamenisch, Y.; Carelli, V.; et al. Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain 2014, 2, 2164–2177. [Google Scholar] [CrossRef] [Green Version]
- Patrick, Y.W.M.; Shankar, S.P.; Biousse, V.; Miller, N.R.; Bean, L.J.; Coffee, B.; Hegde, M.; Newman, N.J. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology 2011, 118, 558–563. [Google Scholar]
- Zerem, A.; Yosovich, K.; Rappaport, Y.C.; Libzon, S.; Blumkin, L.; Liat, B.S.; Lev, D.; Tally, L.S. Metabolic stroke in a patient with bi-allelic OPA1 mutations. Metab. Brain Dis. 2019, 34, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Rizza, T.; Doimo, M.; Legati, A.; Ciolfi, A.; Diodato, D.; Calderan, C.; Carrara, G.; Lamantea, E.; Aiello, C.; et al. Not only dominant, not only optic atrophy: Expanding the clinical spectrum associated with OPA1 mutations. Orphanet J. Rare Dis. 2017, 12, 89. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, J.; Jia, X.; Xiao, X.; Li, S.; Guo, X. Genetic and clinical analyses of DOA and LHON in 304 chinese patients with suspected childhood-onset hereditary optic neuropathy. PLoS ONE 2017, 12, e0170090. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; Den, D.J.T. LOVD v. 2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Schuster, S.; Theurer, Y.; Synofzik, M.; Schöls, L. Generation of optic atrophy 1 patient-derived induced pluripotent stem cells (iPS-OPA1-BEHR) for disease modeling of complex optic atrophy syndromes (Behr syndrome). Stem. Cell Res. 2016, 17, 426–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotto, V.D.; Fogazza, M.; Musiani, F.; Maresca, A.; Aleo, S.J.; Caporali, L.; Morgia, C.; Noll, C.; Lodi, T.; Goffrini, P.; et al. Deciphering OPA1 mutations pathogenicity by combined analysis of human, mouse and yeast cell models. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3496–3514. [Google Scholar] [CrossRef]
- Patrick, Y.W.M.; Chinnery, P.F. Reply: Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e302. [Google Scholar]
- Ferré, M.; Bonneau, D.; Milea, D.; Chevrollier, A.; Verny, C.; Dollfus, H.; Ayuso, C.; Defoort, S.; Vignal, C.; Zanlonghi, X.; et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum. Mutat. 2009, 30, E692–E705. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Barboni, P.; Ross, C.F.N.; Sadun, A.A. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim. Et Biophys. Acta Bioenerg. 2009, 1787, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, D.; Colin, E.; Oca, F.; Ferré, M.; Chevrollier, A.; Guéguen, N.; Valérie, D.D.; Sylvie, N.G.; Barth, M.; Zanlonghi, X.; et al. Letter to the editor: Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Joshi, S.; Singh, K.D.; Kumar, A. Visual evoked potentials: Normative values and gender differences. J. Clin. Diagnostic Res. 2015, 9, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Blazo, M.; Lewis, R.A.; Tonini, R.E.; Takei, H.; Wang, J.; Wong, L.J.; Scaglia, F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol. Genet. Metab. 2011, 103, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Pesch, U.E.A.; Beate, L.K.; Mayer, S.; Jurklies, B.; Kellner, U.; Eckart, A.S.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jung, S.C.; Hong, Y.B.; Yoo, J.H.; Lee, J.H.; Hong, H.D.; Kim, S.B.; Chung, K.W.; Choi, B.O. Recessive optic atrophy, sensorimotor neuropathy and cataract associated with novel compound heterozygous mutations in OPA1. Mol. Med. Rep. 2016, 14, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Carelli, V.; Sabatelli, M.; Carrozzo, R.; Rizza, T.; Schimpf, S.; Wissinger, B.; Zanna, C.; Rugolo, M.; la Morgia, C.; Caporali, L.; et al. ‘Behr syndrome’ with OPA1 compound heterozygote mutations. Brain A J. Neurol. 2015, 138, e321. [Google Scholar] [CrossRef] [Green Version]
- Rubegni, A.; Pisano, T.; Bacci, G.; Tessa, A.; Battini, R.; Procopio, E.; Giglio, S.; Pasquariello, R.; Santorelli, F.M.; Guerrini, R.; et al. Leigh-like neuroimaging features associated with new biallelic mutations in OPA1. Eur. J. Paediatr. Neurol. 2017, 21, 671–677. [Google Scholar] [CrossRef]
- de la Barca, J.M.C.; Delphine, P.M.; Patrizia, A.B.; Ferré, M.; Sarzi, E.; Bris, C.; Leruez, S.; Chevrollier, A.; Desquiret-Dumas, V.; Gueguen, N.; et al. OPA1-related disorders: Diversity of clinical expression, modes of inheritance and pathophysiology. Neurobiol. Dis. 2016, 90, 20–26. [Google Scholar] [CrossRef]
- Milone, M.; Younge, B.R.; Wang, J.; Zhang, S.; Wong, L.J. Mitochondrial disorder with OPA1 mutation lacking optic atrophy. Mitochondrion 2009, 9, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Patrizia, A.B.; Reynier, P.; Layet, V.; Layet, A.; Stevanin, G.; Brissaud, E.; Bonneau, D.; Durr, A.; Brice, A. Heterozygous OPA1 mutations in Behr syndrome. Brain 2011, 134, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.G.; Seto, S.; Ganne, P.; Votruba, M. A randomized, placebo-controlled trial of the benzoquinone idebenone in a mouse model of OPA1-related dominant optic atrophy reveals a limited therapeutic effect on retinal ganglion cell dendropathy and visual function. Neuroscience 2016, 319, 92–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Othman, B.A.; Ong, J.E.; Dumitrescu, A.V. Biallelic Optic Atrophy 1 (OPA1) Related Disorder—Case Report and Literature Review. Genes 2022, 13, 1005. https://doi.org/10.3390/genes13061005
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder—Case Report and Literature Review. Genes. 2022; 13(6):1005. https://doi.org/10.3390/genes13061005
Chicago/Turabian StyleOthman, Bayan Al, Jia Ern Ong, and Alina V. Dumitrescu. 2022. "Biallelic Optic Atrophy 1 (OPA1) Related Disorder—Case Report and Literature Review" Genes 13, no. 6: 1005. https://doi.org/10.3390/genes13061005